
Main-chain liquid crystalline ionomers with a nonplanar ionic segment
Abstract
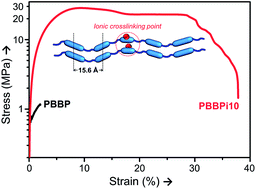
A series of thermotropic main-chain liquid crystalline ionomers named poly(4,4′-bis(6-hydroxyhexyloxy)biphenyl bibenzoate-co-sodium-phenoxaphosphinate-2,8-dicarboxylate) (PBBPins) containing sodium salt of 10H-phenoxaphosphine-2,8-dicarboxylic acid, 10-hydroxy-, 2,8-dimethyl ester, 10-oxide (DMPPO-Na) as an ionic monomer, 4,4′-bis(6-hydroxyhexyloxy)biphenyl (BHHBP) and 4,4′-dicarboxymethylbiphenyl (DBB) as a mesogenic unit were prepared by transesterification. The thermal stability, the mesophase behaviour, and the relationship between the chain structure and thermal properties, as well as the mechanical properties of the main-chain phosphorus-containing liquid crystalline ionomers were investigated. The ionomers all exhibited mesomorphic (smectic A) properties even when the feed ratio of DMPPO-Na : DBB reached as high as 2 : 8 (PBBPi10). In the presence of the physical cross-linking of ionic groups, the viscoelastic behaviour of the ionomers was greatly changed, and mechanical properties were improved with the introduction of an ionic monomer. Thermal stability of the ionomers remained; moreover, residue of the ionomers at 700 °C increased, due to the positive contribution of the phosphorus-containing ionic structure.
 Please wait while we load your content...
Please wait while we load your content...

