
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular self-assembly of novel thermo-responsive double-hydrophilic and hydrophobic Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers†
S.
Petrova
*,
C. G.
Venturini
*,
A.
Jäger
,
E.
Jäger
,
M.
Hrubý
,
E.
Pavlova
and
P.
Štěpánek
Institute of Macromolecular Chemistry, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic. E-mail: petrova@imc.cas.cz; cgventurini@gmail.com; Tel: +420 296 809 322
First published on 15th July 2015
Abstract
Nonlinear amphiphilic block copolymer architectures with precisely controlled structures bring new challenges to biomedical materials research. The paper describes the straightforward synthesis of new “snake tongue“ Y-shaped terpolymers containing poly(ethylene oxide) (PEO), poly(2-ethyl-2-oxazoline) (PEtOx) and poly(ε-caprolactone) (PCL) blocks into structure [AB(C)2] (herein referred to as [MPEO44-b-PEtOx252-b-(PCL)2×44], [MPEO44-b-PEtOx252-b-(PCL)2×87], [MPEO44-b-PEtOx252-b-(PCL)2×131]). A series of well-defined Y-shaped terpolymers were successfully synthesised by a combination of living cationic and anionic ring-opening polymerization (ROP). The selected Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] terpolymer self-assembly was characterised in detail by static and dynamic light scattering, nanoparticle tracking analysis and cryo-transmission electron microscopy. The physico-chemical properties as well as the molecular architecture effect on the self-assembled structures and on the LCST were compared with the Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×87] and the [MPEO44-b-PEtOx252-b-(PCL)2×131] terpolymers. The results indicated a temperature-induced aggregation with an LCST between 60–63 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×44], at 60 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×87] and between 45–50 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×131] with significant differences in the supramolecular self-assembly behaviour compared with the analogous linear structure, clearly indicating the crucial effect of the molecular architecture. Furthermore, the increase of the molecular weight fraction of the hydrophobic block on the Y-shaped triblock terpolymers likely induced a decrease of the LCST.
Introduction
The key feature of successful and versatile polymer materials is the possibility to precisely control the polymer architecture and chemical functionality. Living polymerisation enables the preparation of such polymers with a broad variety of molecular architectures, compositions, side- and end-group functions, as well as the facile preparation of block copolymers with linear and nonlinear architectures.1–7 Star-shaped copolymers are a unique and simple class of macromolecules with a complex architecture, and they constitute a topical area of research due to their intriguing properties, which can be tailored by varying their polymeric chains (arms).Star-shaped block copolymers consisting of at least three linear polymeric arms with a radial arrangement around a central molecular fragment (core)8,9 are usually prepared by the “arm-first” or “core-first” methods. The “arm-first” approach involves the construction of polymer arms on a macroinitiator that contains a precise number of reactive sites.10,11 The “core-first” approach utilises multifunctional low-molecular-weight initiators, allowing for the synthesis of block copolymer chains.12 A method based on difunctional monomers is mentioned in the literature as a third approach for the synthesis of star-shaped copolymers.6 However, this method does not allow for strict control of the number of arms.
It is well known that amphiphilic star-shaped copolymers can easily self-assemble in aqueous media to form nanosized unimolecular micelles containing hydrophobic cores surrounded by hydrophilic shells.13,14 These micellar systems are of great interest for medical uses such as the construction of micellar drug delivery systems.9,15–19 Significant differences in the physicochemical properties of star-shaped copolymers compared with their linear analogues can be observed, such as smaller hydrodynamic volume and, radius of gyration and low melt and solution viscosities, which are beneficial to drug loading and delivery.20–24 It has been shown that linear amphiphilic copolymers have limited applications in drug delivery because they suffer from an initial burst release effect. Especially in systems with non-covalently incorporated drugs, the micellar stability and drug release are difficult to control.25,26 Therefore, various star-shaped copolymers with varying arm numbers and chemical compositions have received considerable attention because of the unique properties and advantages that they possess.27–29 Y-shaped copolymers (typically referred to star copolymers) are another interesting vehicle for drug delivery because they exhibit distinct a micellization behaviour (special stability) compared with the amphiphilic copolymers with a linear architecture.30–32 Furthermore, star copolymers bearing distinct polymeric arms have shown dynamic morphological changes (e.g., micelle-to-unimer transition under certain conditions) because the constituting polymeric components could be designed to be individually responsive to external stimuli such as pH, temperature and solvent.7,33–36 Moreover, a particular subject of even greater interest is the study of biocompatible thermo-responsive self-assembled polymer micelles of amphiphilic, double-hydrophilic and hydrophobic species of star copolymers. It should be noted that the number of such studies is quite limited, and more work is greatly needed to understand the particular characteristics involved in the micellar behaviour of such polymer systems.
In this paper, we describe the synthesis and the study of the self-assembly properties of new “snake tongue” Y-shaped terpolymers based on poly(ethylene oxide) (PEO), poly(2-ethyl-2-oxazoline) (PEtOx) and poly(ε-caprolactone) (PCL) with the general architecture [PEO-b-PEtOx-b-(PCL)2] (Scheme 1). The newly synthesised Y-shaped terpolymers combine environmentally friendly blocks with possible applications in biomedicine. PEtOx was chosen because it exhibits similar chemical and biological properties to PEO;37–39 both polymers are water-soluble and non-toxic.40–43 PEO and PEtOx can be eliminated from the human body if they possess a low enough molar mass. Furthermore, PEtOx in an aqueous solution exhibits a lower critical solution temperature (LCST).44–47 The LCST of PEtOx is ∼61–66.5 °C and strongly depends on the polymer molecular weight (20–500 kDa) and polymer concentration.48,49 PCL is a hydrophobic, nontoxic, biocompatible and fully biodegradable aliphatic polyester.50 To the best of our knowledge, this is the first time that these three blocks were combined in a nonlinear architecture using the “arm-first” method and their supramolecular self-assembly behaviour was compared with analogous blocks of linear architecture and identical weight ratios and molecular weights.
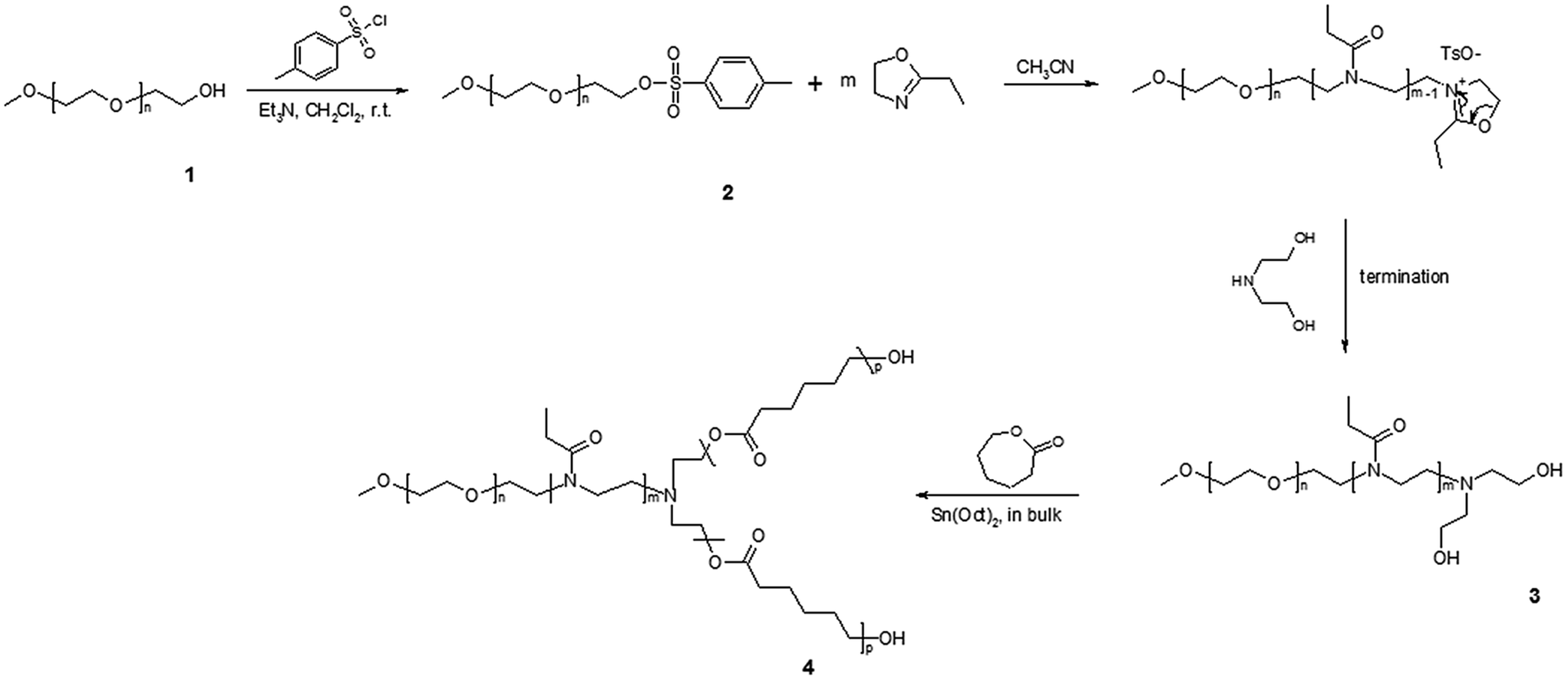 | ||
| Scheme 1 Synthesis of the Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers. | ||
Experimental
Materials
The chemicals were purchased from Sigma-Aldrich Ltd (Prague, Czech Republic). Poly(ethylene oxide monomethyl ether) (MPEO) was used with number-average molecular weight Mn ∼ 1800 g mol−1. The 2-ethyl-2-oxazoline (EtOx, ≥99%) was dried over KOH and CaH2 for 48 h and distilled under dry argon atmosphere before use. ε-Caprolactone (ε-CL, 99%) was dried over CaH2 with continuous stirring at room temperature for 48 h and distilled under reduced pressure before use. Tin(II) bis(2-ethylhexanoate) (Sn(Oct)2, 95%, 0.06 M solution in toluene), p-toluenesulfonyl chloride (≥99%) and diethanolamine (≥98%) were used as received. Triethylamine (TEA, ≥99.5%) was dried over CaH2 and distilled under reduced pressure. Dichloromethane was dried by refluxing over a benzophenone–sodium complex and distilled under argon atmosphere. Toluene (99%) was refluxed for 24 h over CaH2 under dry argon atmosphere and then distilled. Acetonitrile (ACN, 99.8%) was dried by refluxing over CaH2 for 24 h and distilled under argon atmosphere. All other chemicals were used as received.Synthesis of α-methoxy-ω-tosyl-poly(ethylene oxide) macroinitiator ((Stage 2) Scheme 1)
The synthesis of the ω-tosyl-MPEO macroinitiator was carried out according to our previously reported method.51 Yield: 4.25 g, (85%).
1H NMR, δ (TMS, ppm): 2.45 (s, 3H, CH3–), 3.38 (s, 3H, –OCH3), 3.65 (m, 4H, –OCH2CH2–), 4.16 (t, 2H,–CH2O(SO2)), 7.36–7.33 (d, 2H, ArH), 7.82–7.8 (d, 2H, ArH), Mn(NMR) = 2200 g mol−1:
| Mn(SEC) = 2355 g mol−1, Mw/Mn(SEC) = 1.15. |
Synthesis of [MPEO-b-PEtOx(OH)2] diblock copolymer ((Stage 3) Scheme 1)
The polymerisation was carried out as follows; 0.45 g (0.20 mmol) of ω-tosyl-MPEO (Mn(NMR) ∼ 2200 g mol−1) was introduced into a 50 mL glass reactor with a magnetic bar. The macroinitiator was dissolved in dry toluene (15 mL), the solvent was evaporated and the azeotropic drying procedure was twice more repeated. Dry ACN (20 mL) was transferred to the glass reactor using a flame-dried and argon-flushed glass syringe equipped with a metallic cannula. The solution was heated to 80 °C before to inject rapidly, through a septum, 5 mL (49.5 mmol) of 2-ethyl-2-oxazoline, and the polymerisation was continued for five days. To terminate the polymerisation, diethanolamine (0.025 mL, 0.26 mmol, 1.3 eq.) was added and the reaction mixture was stirred for two more hours at 80 °C. Then, the crude product was precipitated in cooled diethyl ether, filtered off, washed with diethyl ether and dried under vacuum overnight at 40 °C. Yield: 4.85 g, (93%).Synthesis of Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers ((Stage 4) Scheme 1)
Typically, 0.5 g (0.162 mmol) of [MPEO-b-PEtOx(OH)2] diblock copolymer (Mn(NMR) ∼![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25750 g mol−1) was introduced into a 50 mL glass reactor with a magnetic bar. The macroinitiator was dissolved in dry toluene and dried three times by azeotropic distillations. A certain amount of freshly distilled ε-CL was then added. After heating to 110 °C a 0.1 mL of 0.06 M Sn(Oct)2 was rapidly injected through a septum and the polymerisation was carried out for 48 h at 110 °C. The reactor was cooled to room temperature and the reaction mixture was dissolved in toluene. The Y-shaped terpolymer was collected by precipitation in cooled diethyl ether, filtrated, and dried overnight at 40 °C in vacuum.
25750 g mol−1) was introduced into a 50 mL glass reactor with a magnetic bar. The macroinitiator was dissolved in dry toluene and dried three times by azeotropic distillations. A certain amount of freshly distilled ε-CL was then added. After heating to 110 °C a 0.1 mL of 0.06 M Sn(Oct)2 was rapidly injected through a septum and the polymerisation was carried out for 48 h at 110 °C. The reactor was cooled to room temperature and the reaction mixture was dissolved in toluene. The Y-shaped terpolymer was collected by precipitation in cooled diethyl ether, filtrated, and dried overnight at 40 °C in vacuum.
Preparation of the nanoparticles (NPs) solutions
To prepare the Y-shaped terpolymer NPs solutions, 5 mg of polymer was dissolved in 1.25 mL of acetone (40 °C), and the polymer solution was added drop wise to 2.5 mL of pure water under magnetic stirring. The acetone was further removed by evaporation under reduced pressure and the solution was concentrated to 1.25 mL. For the scattering measurements the NPs samples were diluted with phosphate buffer saline pH 7.4 (PBS) to the final concentrations of 2.0, 1.5, 1.0, and 0.5 mg mL−1. All samples were filtered using a Millipore 0.45 μm filter (Millipore®, Czech Republic) before the scattering measurements.characterisation techniques
The characterisation techniques such as proton nuclear magnetic resonance (1H NMR), Fourier transform infrared spectroscopy (FT-IR), size exclusion chromatography (SEC), dynamic (DLS) and static (SLS) light scattering, nanoparticle tracking analysis (NTA), cryo-transmission electron microscopy (cryo-TEM) are described in detail in the ESI Section.†Results and discussion
Synthesis of the α-methoxy-ω-tosyl-poly(ethylene oxide) macroinitiator ((Stage 2) Scheme 1)
MPEO end-capped with a tosyl group was prepared as a macroinitiator by esterification reaction. For this purpose, the ω-hydroxyl end-group of commercially available MPEO (Mn(NMR) ∼ 1800 g mol−1) was reacted with an excess of tosyl chloride using CH2Cl2 as a solvent and TEA as a base (compound 2, in Scheme 1). The macroinitiator was fully characterised by 1H NMR, FT-IR spectroscopy and SEC analysis, which are described in detail in our previous report.51Synthesis of the [MPEO-b-PEtOx(OH)2] diblock copolymer ((Stage 3) Scheme 1)
The living cationic ring-opening polymerisation (CROP) of 2-oxazolines (Ox)s (cyclic imino ether) was first reported in 1966 by four independent research groups.52–55 The CROP of 2-oxazolines can proceed in a “living” manner under appropriate conditions, meaning that neither undesired termination nor chain transfer occurs during the polymerisation. Depending on the nature of the monomer and initiator used, the CROP of (Ox)s can be ionic or covalent.56 The living CROP of 2-oxazolines is a versatile method for the preparation of well-defined poly(2-oxazoline)s (POx)s, whereby both the initiation and termination steps provide the possibility of introducing a variety of functional groups. In the case of polymerisation of 2-ethyl-2-oxazoline (EtOx) using sulphonates as electrophilic initiators, the CROP proceeds via ionic species.57–59 Termination of the CROP of (Ox)s can be achieved by nucleophilic attack on the 5-position of the oxazolinium species, which is the thermodynamically controlled and mostly favoured end-capping reaction. The most widely used nucleophilic terminating agents are aqueous or methanolic sodium hydroxide solutions60,61 and carboxylic acid salts. Additionally, termination can occur by the in situ formation of carboxylic acids salts from the acid and 2,6-dimethylpyridine62,63 and amines.61Here, a [MPEO-b-PEtOx(OH)2] double-hydrophilic block copolymer was synthesised by CROP of EtOx using ω-tosyl-MPEO as a macroinitiator and subsequent in situ end-capping by diethanolamine of the living oxazolinium species converted into ω,ω′-dihydroxyl groups, (compound 3, in Scheme 1). The double-hydrophilic block copolymer was obtained with a MPEO molecular weight of approximately 2200 g mol−1 and a PEtOx block molecular weight of approximately 23550 g mol−1. After purification, the structure of the double-hydrophilic block copolymer was confirmed by 1H NMR and FT-IR spectroscopy. The Mn of [MPEO-b-PEtOx(OH)2] was determined by 1H NMR spectroscopy and SEC. The 1H NMR spectrum of the diblock copolymer (Fig. 1) showed the characteristic signals of the protons belonging to the ethylene oxide (EO) and EtOx repeat units. The methylene protons of the EO repeat units marked with b were observed at δ = 3.64 ppm. The spectrum also showed a broad singlet signal observed at δ = 3.46 ppm and labelled as d that corresponded to the chemical shifts of the protons in –N–CH2–CH2 from the EtOx repeat units. Furthermore, the spectrum detected signals corresponding to the pendant group of the main polymer chain of PEtOx at δ = 2.40 ppm (e) that was attributed to the methylene protons of N–C(O)–CH2–CH3 and another labelled as f at δ = 1.12 ppm that was assigned to the methyl group of N–C(O)–CH2–CH3. The 1H NMR spectrum also showed a signal identified as c at δ = 3.70 from the last monomer unit of PEO that was connected to the PEtOx polymer chain. A triplet signal at δ = 2.84 ppm, which corresponded to the four protons from the N–CH2–CH2–HO end-capped group (g and g′) that formed after termination by diethanolamine, was observed. Nevertheless, the signals of the four –CH2–OH (h and h′) hydrogen nuclei were not detectable because they were hidden by the signal of the CH2O units of the PEO repeat unit at δ = 3.64 ppm. Furthermore, the singlet signal at δ = 3.39 ppm attributed to CH3–O– was not detected in the spectrum because it was hidden by the peak of the –N–CH2–CH2– units of the PEtOx repeat unit at δ = 3.46 ppm.
 | ||
| Fig. 1 1H NMR spectrum of the [MPEO-b-PEtOx(OH)2] diblock copolymer in CDCl3 (1, Table 1). | ||
Based on the molecular weight of the initiating ω-tosyl-MPEO macroinitiator, the number-average molecular weight Mn(NMR) of the diblock copolymer was calculated by eqn (1).
| Mn(NMR)[MPEO-b-PEtOx(OH)2] = [(Id/4)/(Ib/4)] × DPMPEO × 99 + Mn(NMR) (macroinitiator) | (1) |
| No | Sample | M n a, (theor.) | M n b, (NMR) | M n c, (SEC) | M w/Mnd, (SEC) | Weight fraction MPEO | Weight fraction PEtOx | Weight fraction PCL |
|---|---|---|---|---|---|---|---|---|
| a M n = [M]o/[I]o × 99 + Mn(NMR) (macroinitiator) and Mn = [M]o/[I]o × 114 + Mn (double-hydrophilic block copolymer). b M n was calculated by 1H NMR spectroscopy according to eqn (1) and (2). c M n values relative to linear PS standards. d M w/Mn values relative to linear PS standards. | ||||||||
| 1 | [MPEO44-b-PEtOx252-(OH)2] | 27200 |
25750 |
30400 |
1.24 | |||
| 2 | [MPEO44-b-PEtOx252-b-(PCL)2×44] | 37200 |
39200 |
44500 |
1.38 | 0.052 | 0.68 | 0.27 |
| 3 | [MPEO44-b-PEtOx252-b-(PCL)2×87] | 47200 |
48600 |
50700 |
1.39 | 0.04 | 0.53 | 0.42 |
| 4 | [MPEO44-b-PEtOx252-b-(PCL)2×131] | 57200 |
59200 |
63800 |
1.33 | 0.034 | 0.44 | 0.53 |
The structure of the obtained diblock copolymer was also confirmed by FT-IR spectroscopy (Fig. 2). The spectrum showed the absorption peaks characteristic of both components (MPEO and PEtOx): at 1625 cm−1 corresponding to the -amide bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) of the 2-ethyl-2-oxazoline repeat units of the PEtOx block; at 1421 cm−1 (δ CH from the CH3 of POx); at 1197 cm−1 attributed to the -ether bond (C–O–C stretching) of the EO repeat units of the PEO backbone; at 1051 cm−1 (C–N stretching from POx); and at 2976 and 2885 cm−1 corresponding to the –C–H vibrations typical for both monomer units. The presence of hydroxyl groups at the ω and ω′ positions in the double-hydrophilic block copolymer structure was proven by the appearance of a broad and intense absorption band with a maximum at 3442 cm−1. Furthermore, a low-intensity absorption band at 3739 cm−1 indicating the existence of intramolecular hydrogen bonding between the hydroxyl groups, was observed.
O stretching) of the 2-ethyl-2-oxazoline repeat units of the PEtOx block; at 1421 cm−1 (δ CH from the CH3 of POx); at 1197 cm−1 attributed to the -ether bond (C–O–C stretching) of the EO repeat units of the PEO backbone; at 1051 cm−1 (C–N stretching from POx); and at 2976 and 2885 cm−1 corresponding to the –C–H vibrations typical for both monomer units. The presence of hydroxyl groups at the ω and ω′ positions in the double-hydrophilic block copolymer structure was proven by the appearance of a broad and intense absorption band with a maximum at 3442 cm−1. Furthermore, a low-intensity absorption band at 3739 cm−1 indicating the existence of intramolecular hydrogen bonding between the hydroxyl groups, was observed.
 | ||
| Fig. 2 FT-IR spectrum of the [MPEO-b-PEtOx(OH)2] diblock copolymer. | ||
The SEC analysis of the [MPEO-b-PEtOx(OH)2] diblock copolymer (bold line, Fig. 3) (1, Table 1) showed a monomodal and narrow molecular weight distribution. The main molecular characteristics of the double-hydrophilic block copolymer are listed in Table 1. The obtained data reported in Table 1 confirmed that the polymerisation was controlled with a low polydispersity index and an experimental molecular weight dictated by the monomer-to-initiator ratio and monomer conversion.
 | ||
| Fig. 3 SEC chromatograms in DMF of the [MPEO-b-PEtOx(OH)2] diblock copolymer (bold line) (1, Table 1) and the Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers (dotted line) (2, Table 1), (dashed line) (3, Table 1), (dash dotted line) (4, Table 1). | ||
Synthesis of the Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymer ((Stage 4) Scheme 1)
In the last step, the double-hydrophilic block copolymer was used as an efficient macroinitiator for the ROP of ε-CL in the presence of Sn(Oct)2 as a catalyst (compound 4, Scheme 1). Different ratios of ε-CL/macroinitiator were used to obtain Y-shaped terpolymers with different PCL block molecular weights, whereas the molecular weight of the double-hydrophilic block copolymer (Mn ∼ 25750 g mol−1) was constant. The composition, structure and molecular weight of the obtained Y-shaped terpolymers were characterised by 1H NMR spectroscopy and SEC analysis. The 1H NMR spectrum of the Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymer (Fig. 4) showed signals typical of PEO, PEtOx and PCL chains. The 1H NMR spectrum showed two singlet signals corresponding to the methylene protons of both the EO and EtOx repeat units at δ = 3.63 and δ = 3.44 ppm marked as b and c, respectively. The characteristic methylene protons of PCL appeared at δ = 4.04 (l) –CH2–OH, δ = 1.63 (i+k) –C(O)–CH2–CH2–CH2–CH2– and δ = 1.36 (j) ppm –C(O)–CH2–CH2–CH2–. The signals of the other protons of the PCL block ((h) –C(O)–CH2– at δ = 2.29 ppm) and the methylene protons of N–C(O)–CH2–CH3 labelled as d at δ = 2.39 ppm corresponding to the pendant group of the PEtOx block overlapped each other. Furthermore, a broad signal in the spectrum was observed and labelled as e at δ = 1.10 ppm, which was assigned to the methyl group –N–C(O)–CH2–CH3 also from the side group of the PEtOx polymer chain. In addition to the characteristic signals of the Y-shaped terpolymer, methylene protons at δ = 2.75 (f) ppm from N–CH2–CH2–OC(O) and at δ = 4.18 (g) ppm from N–CH2–CH2–OC(O) were observed. On the assumption that each macromolecule contained one MPEO, one PEtOx, and one PCL block, the number-average molecular weight of the Y-shaped [AB(C)2] terpolymers should fit eqn (2).| Mn(NMR)[MPEO-b-PEtOx-b-(PCL)2] = [(Il/2)/(Ib/4)] × DPMPEO × 114 + Mn(NMR)[MPEO-b-PEtOx(OH)2] | (2) |
 | ||
| Fig. 4 1H NMR spectrum of the Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymer in CDCl3 (2, Table 1). | ||
The molecular weights and polydispersity indices of the synthesised Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers were determined by SEC. As evidenced the SEC curves were overlapped (see the dotted, dashed and dash dotted lines, Fig. 3). The SEC traces revealed relatively narrow molecular weight distributions commonly observed for “living” controlled polymerisation techniques. The SEC profiles of the Y-shaped terpolymers were monomodal, and the elution times were shifted towards lower values corresponding to higher molecular weights compared with those of the diblock copolymer macroinitiator (bold line, Fig. 3). A slight asymmetry was observed at longer elution time in the SEC curve (dashed line, Fig. 3), which strongly suggested that some unreacted double-hydrophilic block copolymer was present. The main characteristic molecular features of the Y-shaped [AB(C)2] terpolymers are listed in Table 1.
Self-assembly of the Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] terpolymer at 25 °C
The Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] terpolymer was selected (vide Experimental part) to prepare nanoparticle solutions in PBS with a pH of 7.4 (0.5, 1.0, 1.5, and 2.0 mg mL−1). Fig. 5a shows the distribution curves of the hydrodynamic radii (RH) of the nanoparticles measured at a scattering angle of 90°. The diameter distribution (2RH) and relative particle size intensity by NTA are displayed in Fig. 5b and c. The nanoparticles showed a mean particle diameter of 132 nm, and the values of d(0.1), d(0.5), and d(0.9) were 81, 134 and 176 nm, respectively. | ||
Fig. 5 (a) The distribution of the hydrodynamic radii for [MPEO44-b-PEtOx252-b-(PCL)2×44] NPs in PBS solutions (pH 7.4) at concentrations of ( ) 0.5 mg mL−1, ( ) 0.5 mg mL−1, ( ) 1.0 mg mL−1, ( ) 1.0 mg mL−1, ( ) 1.5 mg mL−1, and ( ) 1.5 mg mL−1, and ( ) 2.0 mg mL−1; (b) the distribution of the particle diameter in function of particles concentration, and (c) the distribution of particle diameter in function of the relative intensity. ) 2.0 mg mL−1; (b) the distribution of the particle diameter in function of particles concentration, and (c) the distribution of particle diameter in function of the relative intensity. | ||
The DLS data showed a single population of nanoparticles for each of the four solution concentrations. The size distributions of the particles were narrow, as indicated by NTA that displayed a span value of 0.7 (eqn (4) see ESI†). The RH values (Table 2) were calculated using eqn (1) (see ESI†) through the diffusion coefficient values obtained from the slope of the linear fits of the relaxation rate dependence on q2 (Fig. S1†). The RG parameter was calculated from the linear fit of the angular dependence of Kc/R(θ) based on the data in Fig. 6.
 | ||
Fig. 6 Dependence of Kc/R(θ) on q2 for [MPEO44-b-PEtOx252-b-(PCL)2×44] NPs at solution concentrations of ( ) 0.5 mg mL−1, ( ) 0.5 mg mL−1, ( ) 1.0 mg mL−1, ( ) 1.0 mg mL−1, ( ) 1.5 mg mL−1, and ( ) 1.5 mg mL−1, and ( ) 2.0 mg mL−1. ) 2.0 mg mL−1. | ||
The diffusion coefficient (D) values and the RG/RH ratios were similar (Table 2) for the different concentrations (0.5 to 2.0 mg mL−1). At 25 °C, the D values were constant regardless of the angle of observation, which was in agreement with the scattering contribution of spherical structures.64 The RG/RH ratio, a sensitive structural parameter of the particles in solution,65,66 agreed with a concept of spherical micellar shape in the [MPEO44-b-PEtOx252-b-(PCL)2×44] NP solutions at room temperature. In our previous results,51 a linear triblock terpolymer (MPEO44-b-PEtOx263-b-PCL87) presented similar RH and RG values. Therefore, the resulting assemblies in aqueous media are expected to be related to diffuse core-shell-like or soft ball structures since the hydrophilic (MPEO-b-PEtOx diblock copolymer) corona is much longer then the hydrophobic PCL core. In comparison to the size of the particle shell a small and diffuse core might be expected. Such particles are characterised by high amounts of water entrapped inside the assemblies, lower densities and RG/RH values similar to those obtained in our experiments (RG/RH ∼ 1.0).51,67–71
Self-assembly behaviour at different temperatures
To study the effect of the temperature on the different parameters of [MPEO44-b-PEtOx252-b-(PCL)2×44] in solution (PBS 7.4), dynamic (Fig. S3–5†) and static (Fig. S6 and 7†) light scattering measurements were performed at concentrations from 0.5 to 2.0 mg mL−1. The Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×87] and [MPEO44-b-PEtOx252-b-(PCL)2×137] terpolymers were studied at the concentration of 2 mg mL−1 to compare the physico-chemical properties of the polymer as well as the molecular architecture effect on the self-assembly and on the LCST. Fig. S8† shows the distribution of the hydrodynamic radii for the three synthesised terpolymers NPs in PBS solutions (pH 7.4) at concentrations of 2.0 mg mL−1. Tables 3–5 show respectively the physico-chemical parameters of the [MPEO44-b-PEtOx252-b-(PCL)2×44], [MPEO44-b-PEtOx252-b-(PCL)2×87] and [MPEO44-b-PEtOx252-b-(PCL)2×131] NP solutions obtained from light scattering measurements. The RH values (Tables 3–5, Fig. S8†) were calculated (2 mg mL−1) using eqn (1) (see ESI†), and the diffusion coefficient values were obtained from the slope of the linear fits of the relaxation rate dependence on q2 (Fig. S2†).| T (°C) | R H (nm) | R G (nm)a | R G/RH | M w(NP) (107 g mol−1)a | ρ (g mL−1)b | N agg c |
|---|---|---|---|---|---|---|
| a All the SLS data were obtained from the data in Fig. S6 and 7. b NP density (ρ) was calculated by eqn (3) see ESI. c The aggregation number (Nagg) was calculated by Nagg = Mw(NP)/Mw(unimer). | ||||||
| 5 | 73 | 71 | 0.97 | 1.55 | 0.016 | 386 |
| 15 | 73 | 71 | 0.97 | 1.56 | 0.016 | 389 |
| 25 | 70 | 68 | 0.97 | 1.53 | 0.018 | 382 |
| 40 | 67 | 62 | 0.92 | 1.40 | 0.019 | 349 |
| 45 | 67 | 62 | 0.92 | 1.64 | 0.022 | 410 |
| 50 | 69 | 67 | 0.97 | 1.85 | 0.022 | 462 |
| 55 | 72 | 81 | 1.12 | 2.11 | 0.022 | 527 |
| 60 | 72 | 80 | 1.11 | 4.72 | 0.050 | 1177 |
| 62 | 82 | 134 | 1.63 | 8.73 | 0.063 | 2179 |
For the [MPEO44-b-PEtOx252-b-(PCL)2×44] the DLS data (Fig. S3 and 4†) showed only one population in all of the concentrations up to 55 °C. The zeta potential values for the nanoparticle solutions (2 mg mL−1) varied from −2 to −6 mV (5–70 °C) showing that the values did not change as a function of the temperature. The RG/RH ratio values (0.92–0.97) indicated the presence of structures corresponding to spherical nanoparticles at temperatures up to 50 °C. In addition, nearly constant molecular weight values (1.53–1.85 × 107 g mol−1) and aggregation numbers (349–462) suggested that no particle aggregation was observed in this temperature range (5 to 50 °C). For the [MPEO44-b-PEtOx252-b-(PCL)2×87] (Table 4) the DLS data showed only one scattering population for all temperatures up to 60 °C (data not shown). The zeta potential values for the nanoparticle solutions varied from −4 to −8 mV (5–60 °C) and as for the [MPEO44-b-PEtOx252-b-(PCL)2×44] did not change as a function of the temperature. According to the RG/RH ratio values (0.86–0.97) the presence of structures corresponding to spherical nanoparticles at temperatures up to 50 °C are expected. In addition, nearly constant molecular weight values (0.60–0.74 × 107 g mol−1) and aggregation numbers (118–132) suggested that no particle aggregation was observed in this temperature range (5 to 50 °C). For the last synthesised [MPEO44-b-PEtOx252-b-(PCL)2×131] terpolymer (Table 5) the DLS data showed only one population in all of the concentrations up to 40 °C. The zeta potential values for the nanoparticle solutions varied from −2 to −10 mV (5–50 °C). For these terpolymers the RG/RH ratio values (0.97–1.06) indicated the presence of structures corresponding to spherical nanoparticles at temperatures up to 40 °C. The molecular weight values (0.13–0.21 × 107 g mol−1) and aggregation numbers (20–33) suggested no particle aggregation in the temperature range of 5 to 40 °C.
| T (°C) | R H (nm) | R G (nm)a | R G/RH | M w(NP) (107 g mol−1)a | ρ (g mL−1)b | N agg c |
|---|---|---|---|---|---|---|
| a All the SLS data were obtained from the data in Fig. S6 and 7. b NP density (ρ) was calculated by eqn (3) see ESI. c The aggregation number (Nagg) was calculated by Nagg = Mw(NP)/Mw(unimer). d Sample precipitation. | ||||||
| 5 | 33 | 32 | 0.97 | 0.67 | 0.073 | 132 |
| 15 | 37 | 32 | 0.86 | 0.72 | 0.061 | 142 |
| 25 | 37 | 34 | 0.92 | 0.61 | 0.048 | 120 |
| 40 | 37 | 32 | 0.86 | 0.60 | 0.047 | 118 |
| 45 | 37 | 36 | 0.97 | 0.63 | 0.050 | 124 |
| 50 | 38 | 35 | 0.92 | 0.65 | 0.047 | 128 |
| 55 | 38 | 41 | 1.08 | 0.78 | 0.052 | 154 |
| 60 | 45 | 48 | 1.07 | 1.15 | 0.051 | 227 |
| 62d | — | — | — | — | — | — |
| T (°C) | R H (nm) | R G (nm)a | R G/RH | M w(NP) (107 g mol−1)a | ρ (g mL−1)b | N agg c |
|---|---|---|---|---|---|---|
| a All the SLS data were obtained from the data in Fig. S6 and 7. b NP density (ρ) was calculated by eqn (3) see ESI. c The aggregation number (Nagg) was calculated by Nagg = Mw(NP)/Mw(unimer). d Sample precipitation. | ||||||
| 5 | 63 | 67 | 1.06 | 0.13 | 0.0021 | 20 |
| 15 | 69 | 67 | 0.97 | 0.14 | 0.0017 | 22 |
| 25 | 69 | 73 | 1.06 | 0.16 | 0.0020 | 25 |
| 40 | 77 | 81 | 1.05 | 0.21 | 0.0018 | 33 |
| 45 | 87 | 102 | 1.17 | 0.29 | 0.0017 | 45 |
| 50 | 115 | 132 | 1.15 | 0.32 | 0.0008 | 50 |
| 55d | — | — | — | — | — | — |
| 60d | — | — | — | — | — | — |
| 62d | — | — | — | — | — | — |
At temperatures below the LCST, hydrogen bonds between the polymer carbonyl group and the water hydrogens were abundant,72 and the NPs were swollen by water. The density values confirmed this swollen state, which was also previously verified for linear MPEO44-b-PEtOx263-b-PCL87 NPs.51
When the temperature approached 55 °C for [MPEO44-b-PEtOx252-b-(PCL)2×44] and [MPEO44-b-PEtOx252-b-(PCL)2×87] as well as 40 °C for [MPEO44-b-PEtOx252-b-(PCL)2×131], a slight increase in the values of the RG/RH ratio, molecular weight and aggregation number were observed for the nanoparticles (Tables 3–5). The temperature dependence behaviour observed for the 3 terpolymer NPs was related to the thermodynamic effects of the LCST on the PEtOx block.66 This result was observed because with the increase in temperature, the hydrogen bonds between the carbonyl (CO) group of the PEtOx block and the water hydrogens were weakened.73 The weakening of the hydrogen bonds favoured the expulsion of the water entrapped inside of the particles. Therefore, with an increase in the temperature, an increase in the particle interaction was expected due to the increase in its hydrophobicity. According the results, the onset of NP aggregation driven by PEtOx dehydration started to be observed at ∼55 °C by DLS/SLS measurements for the [MPEO44-b-PEtOx252-b-(PCL)2×44] at ∼50–55 for the [MPEO44-b-PEtOx252-b-(PCL)2×87] and at ∼40 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×131]. Although the increases observed for the NP molecular weight and aggregation number starting from ∼55 °C, [MPEO44-b-PEtOx252-b-(PCL)2×44], ∼50–55, [MPEO44-b-PEtOx252-b-(PCL)2×87], and at 40 °C, [MPEO44-b-PEtOx252-b-(PCL)2×131], were strong indicators of NP aggregation, the RG/RH ratio values also provided valuable information related to this process.64 Commonly, in the aggregated state, the increase in the RG of the NPs is more pronounced in comparison with RH; thus, an increase in the RG/RH ratio is observed. According to previous observations, this behaviour is related to a shift in the position of the scattering centres between the non-aggregated and aggregated NPs.51 In the non-aggregated NPs, the polymer density decreases from the centre to the shell, whereas in the aggregated NPs, the density is also relatively high in the periphery. The scattering centres of the aggregated particles are composed of a collection of hydrophobic collapsed particles, whereas they are water swollen in the non-aggregated particles.
Therefore, higher RG/RH ratios would be expected as the aggregation proceeds with the temperature increase. At 60 °C ([MPEO44-b-PEtOx252-b-(PCL)2×44]) and for nanoparticle concentrations of 0.5 and 1.0 mg mL−1, two peaks were observed in the distribution of the hydrodynamic radii (Fig. S4 c†). The scattering intensity related to the largest peak was approximately 450 nm for both concentrations, indicating a collapsed system at 60 °C. At 61 °C, for the nanoparticle concentration of 1.5 mg mL−1, the presence of a scattering intensity peak corresponding to a size larger than 100 nm (∼429 nm; aggregates) (Fig. S4 d†) was observed. For the nanoparticle concentration of 2 mg mL−1, the collapse was observed at 63 °C (Fig. S5†). Another evidence of the onset of aggregation and nanoparticle collapse was the increase in the diffusion coefficient with the increase on the temperature up to 60 °C (1.95–7.48 × 10−8 cm2 s−1; Fig. S2†). However, at 62 °C a decrease (6.67 × 10−8 cm2 s−1) in the diffusion coefficient indicated the increase in the RH and nanoparticles aggregation. The scattering patterns for the [MPEO44-b-PEtOx252-b-(PCL)2×87] and the [MPEO44-b-PEtOx252-b-(PCL)2×131] follow similar trends with the largest peak (or sample precipitation) indicating a collapsed system at 60–62 °C for [MPEO44-b-PEtOx252-b-(PCL)2×87] and at 45–50 °C for [MPEO44-b-PEtOx252-b-(PCL)2×131] (Fig. S9†). Taking into account the aforementioned results, we may infer that the LCST for the Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] nanoparticle solutions was between 60-63 °C, for the [MPEO44-b-PEtOx252-b-(PCL)2×87] was around 60 °C and for the [MPEO44-b-PEtOx252-b-(PCL)2×131] was between 45–50 °C. Our previous results for linear [MPEO44-b-PEtOx263-b-PCL87] nanoparticle solutions showed a LCST slightly lower, at 56–60 °C51 when compared to [MPEO44-b-PEtOx252-b-(PCL)2×44 and to [MPEO44-b-PEtOx252-b-(PCL)2×87]. The LCST for poly(2-ethyl-2-oxazoline) was observed in the range of 61–66.5 °C and it was dependent of the concentration and molecular weight of the polymer.48 Poly(2-ethyl-2-oxazoline) with concentration between 0.5% and 1% (wt %) and molecular weight of 20000 g mol−1 showed a LCST at 66.5 and 64.5 °C, respectively. In the present work the synthesised Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] and [MPEO44-b-PEtOx252-b-(PCL)2×87] with similar poly(2-ethyl-2-oxazoline) molecular weight presented LCST values of ∼60 °C at the same concentration range. Therefore, the presence of a hydrophobic polymer on the triblock terpolymers induced a decrease of the LCST as previously observed.51 The lowest LCST values (between 45–50 °C, Table 5, Fig. S9†) observed for the largest hydrophobic terpolymer synthesised [MPEO44-b-PEtOx252-b-(PCL)2×131] supports the statement. From these results, a schematic representation of the NPs [MPEO44-b-PEtOx252-b-(PCL)2×44] could be designed (Fig. 7).
 | ||
| Fig. 7 Schematic representation for Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] nanoparticles solutions at different temperatures: (a) in a swollen state, (b) in an aggregation state. | ||
Cryo-TEM microscopy (Fig. 8) was also performed on the NPs to verify the possible morphological changes induced by the temperature. For samples measured at 25 °C, spherical NPs with a mean particle size of 70 nm were observed for [MPEO44-b-PEtOx252-b-(PCL)2×44] (Fig. 8a, top) around 50–60 nm for [MPEO44-b-PEtOx252-b-(PCL)2×87] (Fig. 8c, middle) and 100–120 nm for [MPEO44-b-PEtOx252-b-(PCL)2×131] (Fig. 8e, bottom). NPs measured at higher temperatures showed the onset of aggregation (Fig. 8b, top) for [MPEO44-b-PEtOx252-b-(PCL)2×44] (60 °C) with a mean diameter of 95 nm, around 200 nm (aggregates) for [MPEO44-b-PEtOx252-b-(PCL)2×87] (62 °C) and 300 nm (aggregates) for [MPEO44-b-PEtOx252-b-(PCL)2×131] (55 °C) (Fig. 8e, bottom).
 | ||
| Fig. 8 Cryo-TEM micrographs of polymer NPs prior to temperature changes for [MPEO44-b-PEtOx252-b-(PCL)2×44] (a), [MPEO44-b-PEtOx252-b-(PCL)2×87] (c) and [MPEO44-b-PEtOx252-b-(PCL)2×131] (e) and after temperature increase to 60 °C [MPEO44-b-PEtOx252-b-(PCL)2×44]) (b), to 62 °C [MPEO44-b-PEtOx252-b-(PCL)2×87] (d) and to 55 °C for [MPEO44-b-PEtOx252-b-(PCL)2×131] (f). | ||
The experimental data suggest that the temperature increase caused size and morphological changes in the NPs. The mean diameter of the NPs determined from the cryo-TEM images was in agreement with that determined by DLS (Tables 3–5). It was possible to observe some morphologically ill-defined structures, which were related to inorganic salts (NaCl and KCl) presented in the saline buffer solution.
Conclusions
A series of nonlinear, amphiphilic, biocompatible, Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers with different molecular weights of the PCL block were successfully synthesised by a combination of living cationic and anionic ROP in a three-step synthetic procedure. The ω-tosyl-MPEO macroinitiator was first synthesised using an esterification reaction with tosyl chloride. In a second step, a [PEO-b-PEtOx(OH)2] diblock copolymer was designed by cationic ROP of EtOx using the pre-synthesised ω-tosyl-MPEO macroinitiator. The CROP was terminated in situ by diethanolamine in order to obtain two symmetrical hydroxyl groups (a primary alcohol), which were prone to initiate the anionic ROP of ε-CL catalysed by Sn(Oct)2, providing the final Y-shaped [AB(C)2] terpolymers. The terpolymers were synthesised with good control over the molecular characteristics of each product and with the capability of easily tuning the length of each block. The aqueous self-assembly of the “snake tongue” Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×44] terpolymer was characterised by SLS, DLS, NTA, and cryo-TEM and its physico-chemical properties as well as the molecular architecture effect on the self-assembly and on the LCST was compared with the Y-shaped [MPEO44-b-PEtOx252-b-(PCL)2×87] and [MPEO44-b-PEtOx252-b-(PCL)2×131] terpolymers. The results indicated a temperature-induced aggregation with an LCST between 60–63 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×44], at 60 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×87] and between 45–50 °C for the [MPEO44-b-PEtOx252-b-(PCL)2×131] with significant differences in the supramolecular self-assembly behaviour compared with the analogous linear structure, clearly indicating the crucial effect of the molecular architecture. Furthermore, in agreement with previous findings a decrease in the LCST was observed with increase of the molecular weight fraction of the hydrophobic block (PCL) of the Y-shaped terpolymers.Acknowledgements
This research was supported by the Ministry of Education Youth and Sports of the Czech Republic (LH14079). C.G.V. acknowledges the Program Science without Borders CAPES/Brasil (Process number 2293/13-7). E.J. and A.J. acknowledge Charles University (Prague, CZ) for the financial support and for the opportunity to pursue their PhD studies. The authors acknowledge Eng. Tiago Beck for the drawings of the nanoparticles schematic representations. The research leading to these results has received funding from the Norwegian Financial Mechanism 2009-2014 under Project contract no. 7F14009.References
- K. Matyjaszewski, P. J. Miller, E. Fossum and J. Nakaqwa, Appl. Organomet. Chem., 1998, 12, 667 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and J. Gnanou, Macromolecules, 1998, 31, 7218 CrossRef CAS.
- K. Matyjaszewski, P. J. Miller, J. Pyun, G. Kickelbick and S. Diamanti, Macromolecules, 1999, 32, 6526 CrossRef CAS.
- A. Heise, J. L. Hedrick, M. Trollsas, R. D. Miller and C. W. Frank, Macromolecules, 1999, 32, 231 CrossRef CAS.
- K. Matyjaszewski, Polym. Int., 2003, 52, 1559 CrossRef CAS PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS PubMed.
- R. Riva, W. Lazzari, L. Billiet, F. Du Prez, C. Jérôme and P. Lecomte, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1552 CrossRef CAS PubMed.
- A. Guo, G. Liu and J. Tao, Macromolecules, 1996, 29, 2487 CrossRef CAS.
- S. Petrova, I. Kolev, S. Miloshev, M. D. Apostolova and R. Mateva, J. Mater. Sci.: Mater. Med., 2012, 23, 1225 CrossRef CAS PubMed.
- J. P. Kennedy and S. Jacob, Acc. Chem. Res., 1998, 31, 835 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747 CrossRef CAS PubMed.
- O. A. Matthews, A. N. Shipway and J. F. Stoddart, Prog. Polym. Sci., 1998, 23, 1 CrossRef CAS.
- S. Petrova, R. Riva, C. Jérôme, P. Lecomte and R. Mateva, Eur. Polym. J., 2009, 45, 3442 CrossRef CAS PubMed.
- F. Quaglia, L. Ostacolo, G. Nese, M. Canciello, G. de Rosa, F. Ungaro, R. Palumbo, I. M. La Rotonda and G. Maglio, J. Biomed. Mater. Res., Part A, 2008, 87, 563 CrossRef PubMed.
- K. Knop, M. G. Pavlov, T. Rudolph, K. Martin, D. Pretzel, O. B. Jahn, H. D. H. Scharf, A. A. Brakhage, V. Makarov, U. Möllmann, H. F. Schacherab and S. U. Schubert, Soft Matter, 2013, 9, 715 RSC.
- F. Wang, K. T. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397 CrossRef CAS PubMed.
- C.-L. Peng, M.-J. Shieh, M.-H. Tsai, C.-C. Chang and P.-S. Lai, Biomaterials, 2008, 29, 3599 CrossRef CAS PubMed.
- J. Cheng, J.-X. Ding, Y.-C. Wang and J. Wang, Polymer, 2008, 49, 4784 CrossRef CAS PubMed.
- F. C. Giacomelli, P. Stepanek, C. Giacomelli, V. Schmidt, E. Jäger, A. Jäger and K. Ulbrich, Soft Matter, 2011, 19, 9316 RSC.
- B. S. Lele and J.-C. Leroux, Polymer, 2002, 43, 5595 CrossRef CAS.
- G. Lapienis, Prog. Polym. Sci., 2009, 34, 852 CrossRef CAS PubMed.
- F. Sanda, H. Sanada, Y. Shibasaki and T. Endo, Macromolecules, 2002, 35, 680 CrossRef CAS.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453 CrossRef CAS.
- C. A. P. Joziasse, H. Grablowitz and A. J. Pennings, Macromol. Chem. Phys., 2000, 201, 107 CrossRef CAS.
- J. G. Ryu, Y. I. Jeong, I. S. Kim, J. H. Lee, J. W. Nah and S. H. Kim, Int. J. Pharm., 2000, 200, 231 CrossRef CAS.
- A. Layre, P. Couvreur, H. Chacun, J. Richard, R. C. Passirani, D. Requier, J. P. Benoit and R. Gref, J. Controlled Release, 2006, 111, 271 CrossRef CAS PubMed.
- Y. Y. Yuan, Y. C. Wang, J. Z. Du and J. Wang, Macromolecules, 2008, 41, 8620 CrossRef CAS.
- Y. Q. Zhu and S. P. Gido, Macromolecules, 2003, 36, 5719 CrossRef CAS.
- Z. S. Ge, Y. L. Cai, J. Yin, Z. Y. Zhu, J. Y. Rao and S. Y. Liu, Langmuir, 2007, 23, 1114 CrossRef CAS PubMed.
- H. H. Zhang, Z. Q. Huang, B. W. Sun, J. X. Guo, J. L. Wang and Y. Q. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8131 CrossRef CAS PubMed.
- J. Y. Rao, Y. F. Zhang, J. Y. Zhang and S. Y. Liu, Biomacromolecules, 2008, 9, 2586 CrossRef CAS PubMed.
- J. Sun, X. S. Chen, J. S. Guo, Q. Shi, Z. G. Xie and X. B. Jing, Polymer, 2009, 50(455), 2009 Search PubMed.
- K. van Butsele, F. Stoffelbach, R. Jérôme and C. Jérôme, Macromolecules, 2006, 39, 5652 CrossRef CAS.
- J. Yang, D. Zhang, S. Jiang, J. Yang and J. Nie, J. Colloid Interf. Sci., 2010, 352, 405 CrossRef CAS PubMed.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2008, 24, 12001 CrossRef CAS PubMed.
- J. Song, E. Lee and B.-K. Cho, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 446 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986 CrossRef CAS PubMed.
- A. Mero, G. Pasut, L. Dalla Via, M. W. Fijten, U. S. Schubert, R. Hoogenboom and F. M. Veronese, J. Controlled Release, 2008, 125, 87 CrossRef CAS PubMed.
- O. Sedláček, B. D. Monner, S. K. Filippov, R. Hoogenboom and M. Hrubý, Macromol. Rapid Commun., 2012, 33, 1648 CrossRef PubMed.
- W. H. Velander, R. D. Madurawe, A. Subramanian, G. Kumar, G. Sinai-Zingde, J. S. Riffle and C. L. Orthner, Biotechnol. Bioeng., 1992, 39, 1024 CrossRef CAS PubMed.
- D. D. Lasic and D. Needham, Chem. Rev., 1995, 95, 2601 CrossRef CAS.
- S. Zalipsky, C. B. Hansen, J. M. Oaks and T. M. Allen, J. Pharm. Sci., 1996, 85, 133 CrossRef CAS PubMed.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848 CrossRef CAS PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724 CrossRef CAS PubMed.
- H. Uyama and S. Kobayashi, Chem. Lett., 1992, 21, 1643 CrossRef.
- J. Zhao, R. Hoogenboom, G. van Assche and B. van Mele, Macromolecules, 2010, 43, 6853 CrossRef CAS.
- M. Hrubý, S. K. Filippov, J. Pánek, M. Novakova, H. Mackova, J. Kučka, D. Vetvicka and K. Ulbrich, Macromol. Biosci., 2010, 10, 916 CrossRef PubMed.
- P. Lin, C. Clash, E. M. Pearce, T. K. Kweil and M. A. Aponte, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 603 CrossRef CAS PubMed.
- D. Christova, R. Velichkova, W. Loos, E. J. Goethals and F. Du Prez, Polymer, 2003, 44, 2255 CrossRef CAS.
- J.-G. Ryu, Y.-I. Jeong, I.-S. Kim, J.-H. Lee, J.-W. Nay and S.-H. Kim, Int. J. Pharm., 2000, 200, 231 CrossRef CAS.
- S. Petrova, C. G. Venturini, A. Jäger, E. Jäger, P. Černoch, S. Kereïche, L. Kováčik, I. Raška and P. Štěpánek, Polymer, 2015, 59, 215 CrossRef CAS PubMed.
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 2253 CrossRef CAS PubMed.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Their and H. Hellmann, Angew. Chem., 1966, 20, 913 CrossRef PubMed.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, Polym. Lett., 1966, 4, 441 CrossRef CAS PubMed.
- A. Levy and M. Litt, Polym. Lett., 1967, 5, 871 CrossRef PubMed.
- C. Kim, S. C. Lee, S. W. Kang, I. C. Kwon and S. Y. Jeong, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 2400 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659 CrossRef CAS PubMed.
- M. W. M. Fijten, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4804 CrossRef CAS PubMed.
- B. Brissault, C. Guis and H. Cheradame, Eur. Polym. J., 2002, 38, 219 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151 CrossRef CAS.
- J.-S. Park and K. Kataoka, Macromolecules, 2006, 39, 6622 CrossRef CAS.
- S. Kobayashi, E. Masuda, S. I. Shoda and Y. Shimano, Macromolecules, 1989, 22, 2878 CrossRef CAS.
- M. Miyamoto, K. Naka, M. Tokumizu and T. Saegusa, Macromolecules, 1989, 22, 1604 CrossRef CAS.
- H. Huang, R. Hoogenboom, M. A. M. Leenen, P. Guillet, A. M. Jonas, U. S. Schubert and J.-F. Gohy, J. Am. Chem. Soc., 2006, 128, 3784 CrossRef CAS PubMed.
- W. Burchard, Adv. Polym. Sci., 1983, 48, 1 CrossRef CAS.
- D. Xie, K. Xu, R. Bai and G. Zhang, J. Phys. Chem. B, 2007, 111, 778 CrossRef CAS PubMed.
- C. E. Castro, B. Mattei, K. A. Riske, E. Jäger, A. Jäger, P. Štěpánek and F. C. Giacomelli, Langmuir, 2014, 30, 9770 CrossRef PubMed.
- P. Dimitrov, M. Jamroz-Piegza, B. Trzebicka and A. Dworak, Polymer, 2007, 48, 1866 CrossRef CAS PubMed.
- J. Fu and C. Wu, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 703 CrossRef CAS.
- J. Fu, X. Li, D. K. P. Ng and C. Wu, Langmuir, 2002, 18, 3843 CrossRef CAS.
- L. Yang, X. Qi, P. Liu, A. El Ghzaoui and S. Li, Int. J. Pharm., 2010, 394, 43 CrossRef CAS PubMed.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686 CrossRef CAS PubMed.
- F. P. Chen, A. E. Ames and L. D. Taylor, Macromolecules, 1990, 23, 4688 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08298f |
| This journal is © The Royal Society of Chemistry 2015 |