
Sn–Co nanoparticles encapsulated in grid-shell carbon spheres, applied as a high-performance anode material for lithium-ion batteries
Abstract
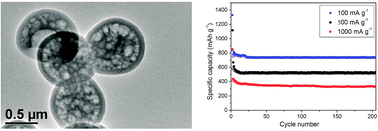
A novel composite comprising Sn–Co nanoparticles encapsulated in grid-shell carbon spheres was synthesized. The composite comprised homogeneous grid-shell carbon spheres with a size of ∼1 μm and a shell thickness of ∼100 nm; SnCo and SnO2 nanoparticles with an average size of ∼16 nm were uniformly embedded in the shells and internal grids of the carbon spheres. This composite showed high capacity, good rate performance and excellent capacity retention when it was applied as an anode material for lithium-ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

