
Au decorated Fe3O4@TiO2 magnetic composites with visible light-assisted enhanced catalytic reduction of 4-nitrophenol
Abstract
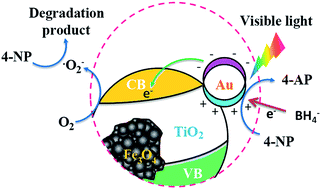
Heterostructured Au nanoparticle decorated Fe3O4@TiO2 composite magnetic microspheres (MSs) were synthesized by grafting Au nanoparticles onto 3-aminopropyltrimethoxysilane (APTMS) modified Fe3O4@TiO2 MSs. Significantly, by varying the reaction conditions, the as-synthesized Fe3O4@TiO2@Au MSs showed high performance in the catalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in the presence of NaBH4 under visible light. In addition, the as-prepared Fe3O4@TiO2@Au MSs can clean themselves by photocatalytic degradation of organic molecules, and can be reused for several cycles with convenient magnetic separability. This approach provided a platform based on the synergy of varying components under suitable conditions to optimize the catalytic ability.
 Please wait while we load your content...
Please wait while we load your content...

