DOI:
10.1039/C5RA08103C
(Paper)
RSC Adv., 2015,
5, 52926-52937
Elucidating structural basis of benzofuran pyrrolidine pyrazole derivatives for enhancing potency against both the InhA enzyme and intact M. tuberculosis cells: a combined MD simulations and 3D-QSAR study†
Received
2nd May 2015
, Accepted 10th June 2015
First published on 10th June 2015
Abstract
A 2-trans enoyl-acyl carrier protein (ACP) reductase or InhA of M. tuberculosis is a drug target of isoniazid (INH), the first-line drug for tuberculosis treatment. Many series of compounds have been developed as novel inhibitors of this enzyme. However, they lack good potency against purified InhA and activity against intact M. tuberculosis cells. Benzofuran pyrrolidin pyrazole derivatives are potent direct InhA inhibitors. These compounds show high potency for InhA inhibition with IC50 values at nanomolar levels. However, their activities against M. tuberculosis cells in terms of MIC90 were about one-thousand fold than IC50. Accordingly, in this work, IC50 and MIC90 values of benzofuran pyrrolidin pyrazole derivatives were subjected to CoMFA and CoMSIA studies in order to investigate the structural basis required for good activity against both purified InhA and M. tuberculosis cells. Moreover, MD simulations were employed to evaluate key interactions for binding benzofuran pyrrolidin pyrazole derivatives in InhA. Based on MD results, the core structure of these compounds is the key portion for binding in the InhA pocket. Alternatively, R substituents showed weak interactions with the InhA pockets. Interpretation of IC50 and MIC90 CoMSIA contour maps revealed the structural requirements in terms of steric, electrostatic, hydrophobic and hydrogen donor and acceptor for IC50 and MIC90 values of InhA inhibitors. Finally, the integrated results obtained from MD simulations and graphic interpretation of CoMSIA models provided a structural concept for rational design of novel InhA inhibitors with better potency against both the InhA enzyme and intact M. tuberculosis cells.
1. Introduction
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (M. tuberculosis) and remains one of the world's deadliest infectious diseases. The World Health Organization (WHO) reported that an estimated 9.0 million people developed new TB cases and 1.5 million people died from this disease in 2013. Moreover, the incidence of new TB cases and deaths in 2013 was higher than those reported previously.1 The high mortality rate of TB is caused by multi drug-resistant tuberculosis (MDR-TB),2–7 extensively drug-resistant tuberculosis (XDR-TB),8,9 totally drug-resistant tuberculosis (TDR-TB)10,11 and human immunodeficiency virus (HIV) co-infection.1 A NADH-dependent 2-trans enoyl-acyl carrier protein (ACP) reductase or InhA has been identified as potential drug target for tuberculosis treatment.12 This enzyme catalyzes the reduction of α,β-unsaturated fatty acids, the last step in fatty acids biosynthesis in M. tuberculosis.12–14 InhA was reported as the drug target of isoniazid (INH), the first-line drug against tuberculosis.15–23 Since INH is a prodrug, it requires the activation process of catalase-peroxidase (KatG) to generate the acyl radical active form. This radical is then covalently bound to nicotinamide adenine dinucleotide (NAD+) to produce an active INH-NAD adduct acting as a potent InhA inhibitor.18–23 The high potency of INH against InhA was lost by mutations in KatG. Therefore, many researchers aimed to discover novel inhibitors that can directly inhibit InhA without the KatG activation process. Inhibitors that can act like this are called direct InhA inhibitors. A class of N-((3R,5S)-1-(benzofuran-3-carbonyl)-5-carbamoylpyrrolidin-3-yl)-1H-pyrazole-5-carboxamide derivatives (benzofuran pyrrolidin pyrazole derivatives) have been identified as potent direct InhA inhibitors.24 The majority of benzofuran pyrrolidine pyrazole derivatives show high potency against purified InhA with inhibitory concentration of compound required to inhibit InhA at 50% (IC50) values at the nanomolar level. However, these compounds show weak cellular activity against M. tuberculosis, with the minimum inhibitory concentration of compound that resulted in complete inhibition in growth of M. tuberculosis 90% (MIC90) at the micromolar level. These results show poor correlation between IC50 and MIC90 values of benzofuran pyrrolidine pyrazole derivatives. In this work, IC50 and MIC90 values of benzofuran pyrrolidine pyrazole derivatives were used for comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) studies in order to investigate the structural basis of these compounds for good activity against both InhA and M. tuberculosis. Moreover, molecular dynamics (MD) simulations were employed to evaluate the key interactions for binding of benzofuran pyrrolidin pyrazole derivatives in InhA. Therefore, the integrated results obtained from MD simulations and graphic interpretation of quantitative structure activity relationship (QSAR) models should provide crucial structural concepts for improving the correlation between IC50 and MIC90 values of benzofuran pyrrolidin pyrazole derivatives.
2. Material and methods
2.1 Data sets and biological activities
Thirty-four benzofuran pyrrolidin pyrazole derivatives used for CoMFA and CoMSIA studies were identified from the published literature.24 Chemical structures and experimental biological activities in terms of MIC90 and IC50 values of these compounds are shown in Table 1. MIC90 and IC50 values were nominally converted into log(1/MIC90) and log(1/IC50) values for CoMFA and CoMSIA studies. Based on the diversity of structures and wide range of activities, the data set of compounds was divided into 30 training set compounds for final model development and 4 test set compounds for model validation. All chemical structures of benzofuran pyrrolidin pyrazole derivatives were constructed using the standard tools available in the GaussView 3.07 program and were then fully optimized using the HF/6-31G method implemented in the Gaussian 09 program.25 The harmonic vibrational frequencies of the optimized geometries have also been calculated. All elements in the calculated Hessian matrix are positive, which indicate that the structures are true minima on the potential energy surface.
Table 1 The chemical structures and activities against InhA and M. tuberculosis of thirty-four benzofuran pyrrolidin pyrazole derivatives
2.2 Molecular docking calculations
In this study, molecular docking calculations using the GOLD Program26–30 were employed with the aims of generating the initial structure for MD simulations and performing molecular alignment to set up CoMFA and CoMSIA models. The available X-ray structure of InhA in a complex with compound 1 (PDB code 4COD) was used as an initial structure for molecular docking calculations. All atoms of the protein were kept rigid, whereas ligand was flexible during the molecular docking calculations. The number of Genetic Algorithm (GA) runs was set to 15 runs with the default search algorithm parameters. The docking calculations were validated using the root-mean-square deviation (RMSD) value between the docked and observed X-ray conformations of compound 1 in its pocket. A RMSD value lower than 1 Å was acceptable. Then, molecular docking calculations with validated parameters were used to dock all remaining compounds into the InhA binding pocket. The binding mode that showed the lowest binding energy was selected for each compound and was used to set up CoMFA and CoMSIA models. It was then used as the initial structure for MD simulations of compounds 2, 22, 23 and 28.
2.3 Molecular dynamics simulations
Compound 28, with the best IC50 value, was selected to investigate its binding mode in InhA. Moreover, the binding modes of compounds 2, 22 and 23 were modelled by MD simulations in order to investigate the effect of R2 and R3 substituents on the IC50 value. The AMBER12 program31 was employed to perform molecular dynamics simulations. The complex structures of compounds 2, 22, 23 and 28 in InhA obtained from molecular docking calculations were used as the initial structure in MD simulations. The Amber ff03 force field was used for the physical description of InhA.32 The general Amber force field (GAFF)33,34 and restrained electrostatic potential (RESP) partial charges35–38 of ligands and NAD+ were generated by the antechamber module implemented in the AMBER12 package. To generate the system for MD simulations, the initial complex structure was solvated by TIP3P water39 in a truncated octahedral box extending up to 10 Å from the solute species. Five Na+ ions were added to neutralize the system charge. Initially, the energy of system was minimized using a steepest decent method followed by the conjugate gradient method. Then, the system was gradually warmed from 0 K to 300 K in 30 ps by restraining all atoms of the complex with a restraint weight of 2 kcal mol−1 Å−2. This was followed by 70 ps of the position-restrained dynamics simulations with a restraining weight of 2 kcal mol−1 Å−2 at 300 K under an isobaric condition. Finally, 10 ns MD simulations without any restraints were performed using the same conditions. Long-range electrostatic interactions were applied using the Particle Mesh Ewald method (PME)40 during the simulations. The cut-off distance for the long-range van der Waals interaction was set to 8 Å. The SHAKE method41 was applied to constrain the bond lengths of hydrogen atoms attached to heteroatoms. Coordinates and energy outputs during MD simulations were recorded at 2 ps intervals.
2.4 Binding free energy calculations
The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM-PBSA) method42–45 was employed for calculating the binding free energy of compounds 2, 22 and 23 in InhA. In this calculation, 250 snapshots of the complex, receptor and ligand were extracted every 8 ps from the last nanosecond of the MD trajectory, which represents the equilibrium state. The binding free energy (ΔGbind) of compounds 2, 22 and 23 complexed with InhA were estimated from eqn (1), where ΔGvacuum and ΔGsolv were the binding free energy of the complex in vacuum and the solvation free energy, respectively. In the MM-PBSA approach, the solvation free energy was calculated by solving a linearized Poisson–Boltzman equation. ΔGvacuum was obtained by calculating the interaction energy between InhA and compounds 2, 22 and 23 (ΔEMM) and taking the entropy change (TΔS) as shown in eqn (2). ΔEMM is divided into three components, non-covalent van der Waals energy (ΔGvdW), electrostatic energy (ΔGele) and internal energy (ΔGint), as shown in eqn (3). ΔEMM and ΔGsolv were calculated using the SANDER module and a PBSA program of the AMBER suite, respectively. The entropy contribution was estimated using normal mode analysis with the NMODE module.46 The entropy contribution was estimated using 250 snapshots for the binding free energy calculation.| | |
ΔGbind = ΔGvacuum + ΔGsolv
| (1) |
| | |
ΔGvacuum = ΔEMM − TΔS
| (2) |
| | |
ΔEMM = ΔGvdW + ΔGele + ΔGint
| (3) |
2.5 CoMFA and CoMSIA methods
IC50 and MIC90 values of compounds were used to set up CoMFA47 and CoMSIA48 models in order to evaluate the key structural features relating to the activity against both InhA and M. tuberculosis. The predicted binding modes of training set compounds obtained from molecular docking calculations were used for molecular alignment to set up CoMFA and CoMSIA models. SYBYL 8.0 molecular modelling software was used to run CoMFA and CoMSIA models. Partial least square (PLS) analysis was employed to derive a linear relationship between CoMFA and CoMSIA descriptor fields and activities. The PLS analysis, using the leave-one-out (LOO) cross-validation method, was performed to determine the optimal number of components. Sequentially, a final analysis with the optimal number of components was performed to construct CoMFA and CoMSIA models that were not cross-validated. The non-cross-validated correlation coefficient (r2) and the leave-one-out cross-validated correlation coefficient (q2) were used to evaluate the predictive ability of CoMFA and CoMSIA models. Selected CoMFA and CoMSIA models were employed to predict IC50 and MIC90 values of test set compounds that were not used to construct models. This was done to evaluate the external predictive ability of these models.
3. Results
3.1 Stability of the complex models
To reveal the structural stability of simulation system, the RMSD values for the position of all solute species were separately analyzed. The RMSD plots for the four simulation systems over 10 ns are shown in Fig. 1. Convergent RMSD plots indicated that the equilibrium state was reached for each system during this simulation period. As shown, the RMSDs for compounds 2, 22, 23 and 28 in InhA converged after approximately 2 ns.
 |
| | Fig. 1 RMSD plots of compounds 2 (a), 22 (b), 23 (c), and 28 (d) complexed with InhA. | |
3.2 Reliability of the calculation methods
MD simulations were employed to model the binding modes of compounds 2, 22, 23 and 28 in the InhA pocket. The experimental binding free energy (ΔGexp) lying within the experimental error of the calculated values (ΔGbind) considered as the correlation between the experimental binding free energy and the calculated values was used to indicate the reliability of the modelled binding modes of these compounds. ΔGbind values of compounds 2, 22, 23 and 28 were close to their ΔGexp values (Table 2). Therefore, we concluded that MD simulations reliably modelled binding modes of compounds 2, 22, 23 and 28 in the InhA pocket.
Table 2 ΔGbind and ΔGexp of compounds 2, 22, 23 and 28 in InhA (kcal mol−1)
| Cpd. |
ΔH |
−TΔS |
ΔGbind |
ΔGexp |
| 2 |
−46.91 ± 5.08 |
−31.03 ± 6.06 |
−15.88 ± 5.14 |
−15.52 |
| 22 |
−49.69 ± 3.87 |
−33.15 ± 6.41 |
−16.54 ± 4.80 |
−15.82 |
| 23 |
−49.61 ± 3.71 |
−32.79 ± 5.57 |
−16.82 ± 4.79 |
−15.65 |
| 28 |
−49.26 ± 4.45 |
−32.52 ± 6.58 |
−16.74 ± 5.34 |
−16.07 |
3.3 Binding mode of compound 28
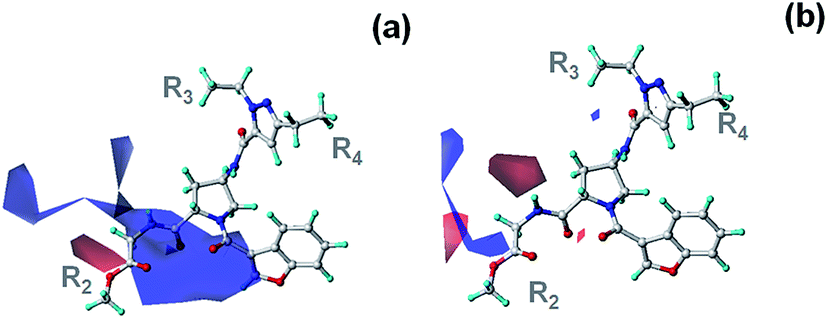
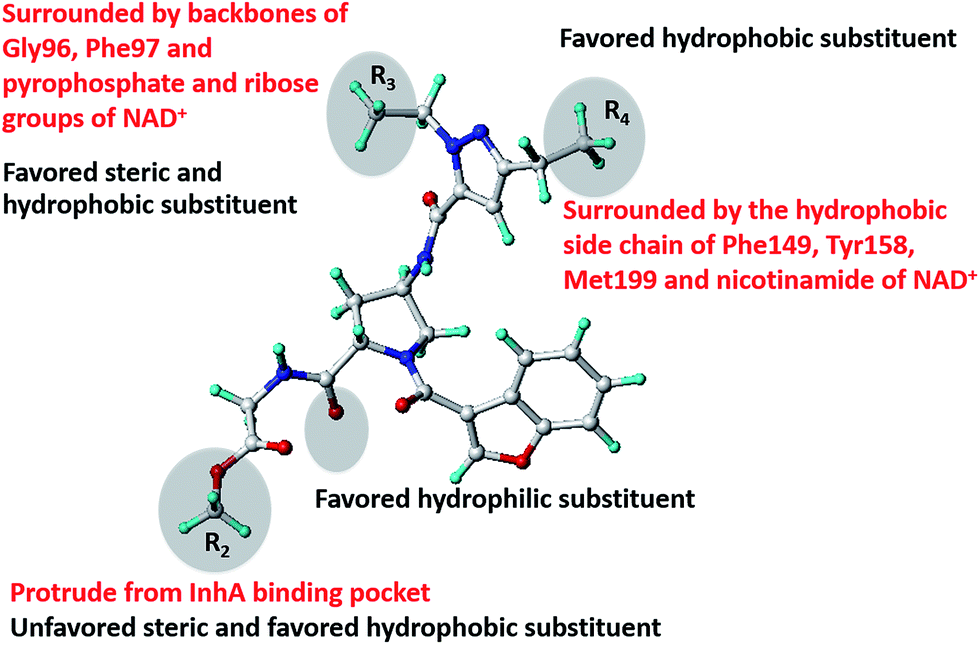
The binding mode of compound 28 complexed with InhA obtained from MD simulations is shown in Fig. 2. Residues located near each substituent and the core structure are listed in Fig. 3. A hydrogen atom (the R1 substituent) is near the carbonyl backbone of Met103. 2-pyridinyl methyl (the R2 substituent) protrudes from the InhA pocket and interacts with the solvent (Fig. 2). The ethyl moiety (the R3 substituent) is located near backbones of Gly96, Phe97 and pyrophosphate and ribose groups of NAD+. The ethyl group (the R4 substituent) was located in the hydrophobic side chains of Phe149, Tyr158, Met199 and nicotinamide of NAD+. With regard to the core structure, the pyrazole ring in the core structure was sandwiched between two hydrophobic side chains of Met161 and Ala198. CO and NH of pyrazole amide formed hydrogen bonds with the backbones of Met98 and Ala198, respectively. The benzofuran core was buried in the hydrophobic side chains of Ile215, Ala157, Ile202 and Ala201, and was sandwiched between the hydrophobic side chains of Leu207 and Met103. The carbonyl of benzofuran core formed a hydrogen bond with the NH backbone of Ala201. NH of pyrrolidine amide formed a hydrogen bond with the CO backbone of Leu197.
 |
| | Fig. 2 Compound 28 (cyan) in its complex with whole InhA (grey) obtained from MD simulations. | |
 |
| | Fig. 3 List of residues surrounding within 4 Å from compound 28. | |
3.4 Interaction energy
Free-energy decomposition calculations were used to investigate the interaction energies between compound 28 and each residue in the InhA pocket. Fig. 4 shows these interaction energies obtained from free-energy decomposition calculations. The lowest interaction energy (−7.42 kcal mol−1) was observed for Met103, indicating that this residue had the largest contribution to binding of compound 28 in the InhA pocket. As previously mentioned, Met103 and Leu207 were sandwiched in the benzofuran core. Another remarkable interaction energy (−7.06 kcal mol−1) was found for NAD+. This was responsible for van der Waal and electrostatic interactions with the R3 and R4 substituents of compound 28 (Fig. 3). Ala198 showed an interaction energy (−6.16 kcal mol−1), comparable with those of Met103 and NAD+. This residue formed hydrogen bonds with the NH of pyrazole amide and sandwiched the pyrazole ring (Fig. 3). Met98, Leu197 and Ala201 formed other hydrogen bonds with the core structure with interaction energies of −2.94, −3.27 and −5.33 kcal mol−1, respectively. Based on interaction energy profile of compound 28, the core structure formed more attractive interactive energies with surrounding residues than R substituents (Fig. 4). This result indicates that the core structure is the key fragment for binding of this compound in the InhA pocket.
 |
| | Fig. 4 Interaction energy profile of compound 28 and surrounding residues within 4 Å. | |
3.5 The effect of the R2 substituent on IC50 and MIC90 values
As compared with the positions of other R substituents, the R2 position had the most varied substituents (Table 1). Compound 28 exposing the 2-pyridylmethyl at the R2 position showed the best activity for InhA inhibition with an IC50 of 0.002 μM. When the R2 substituent of this compound was replaced by CH2COOMe (compound 22), the IC50 value was slightly changed to 0.003 μM. In contrast, the MIC90 value against whole M. tuberculosis cell was greatly changed from 0.7 μM to 0.05 μM (Table 1). To reveal the effect of the R2 substituent on the IC50 value, the binding modes of compounds 28 and 22 were compared (Fig. 5). The binding modes of these compounds in the InhA pocket were similar, and the R2 substituents occupied in the same positions. Moreover, the interaction energy profiles of compounds 28 and 22 with residues in InhA pocket were similar (Fig. 6). As discussed above, the R2 substituent of compound 28 protruded from the InhA pocket leading to weak interaction of this substituent with the pocket. Therefore, the IC50 value against InhA was not significantly changed when the R2 substituent was varied. When the R2 substituent was replaced by a hydrogen atom (compound 23), the binding mode and interaction energy profile of this compound were similar to those of compounds 22 and 28 (Fig. 5 and 6). With regard to IC50 values, compound 23 showed a comparable IC50 value with those of compounds 22 and 28. However, the MIC90 value of this compound (0.5 μM) was largely increased over that of compound 22 (0.05 μM). These results indicate that the R2 substituent had a small effect on the IC50 value against InhA due to its weak interaction with the InhA pocket. Alternatively, this substituent is crucial to controlling the MIC90 against intact M. tuberculosis cells.
 |
| | Fig. 5 The superimposition of binding modes of compounds 22 (pink), 23 (cyan) and 28 (green). | |
 |
| | Fig. 6 Comparison of the interaction energy profiles of compounds 22 (green), 23 (blue) and 28 (yellow) with surrounding pocket within 4 Å. | |
3.6 The effect of the R3 substituent on IC50 and MIC90 values
The R3 substituent of compounds in the data set was varied as ethyl (Et) or methyl (Me) groups (Table 1). Compounds 2 and 22 with structural differences at the R3 substituent were selected to show the effect of the R3 substituent on IC50 and MIC90 values. IC50 values of these compounds (0.005 and 0.003 μM, respectively) were not significant different, but their MIC90 values were tenfold different (0.5 and 0.05 μM, respectively). Fig. 7 shows the binding modes of compounds 2 and 22 in InhA obtained from MD simulations. The R3 substituents of these compounds were located in the same position and surrounded by backbones of Gly96, Phe97 as well as pyrophosphate and ribose groups of NAD+. The ethyl group (The R3 substituent) of compound 22 is close to Phe97 and pyrophosphate and ribose groups of NAD+ more than the methyl group of compound 2. Therefore, interaction energies of compound 22 with Phe97 and NAD+ had greater attraction than those of compound 2 (Fig. 8). Moreover, the presence of a methyl group at the R3 position of compound 2 shifted the position of benzofuran core surrounded by Met103 and Ile202, and disrupted hydrogen bond interaction with Met98. Accordingly, interaction energies of compound 2 with Met98, Met103 and Ile202 showed less attraction than those of compound 22 (Fig. 8). These results indicate that compound 22 should have a better IC50 against InhA compared to compound 2. However, other than the interaction energies of Met98, Met103, Ile202, Phe97 and NAD+, compounds 2 and 22 are comparable. The IC50 value for InhA inhibition by compound 22 was slightly better than that of compound 2. However, its MIC90 value was tenfold better than that of compound 2. The results indicated that the ethyl group at the R3 position is more conducive to favorable IC50 and MIC90 values than the methyl group.
 |
| | Fig. 7 The superimposition of binding modes of compounds 2 (yellow) and 22 (pink). | |
 |
| | Fig. 8 Comparison of the interaction energy profiles of compounds 2 (gray) and 22 (green) with surrounding pocket within 4 Å. | |
3.7 CoMFA and CoMSIA models
In this study, CoMFA and CoMSIA models were constructed from IC50 and MIC90 where prefixed with IC50 and MIC90, respectively. IC50 and MIC90 CoMSIA models were constructed based on various combinations of molecular descriptor fields, in order to develop a highly predictive CoMSIA model (Tables 3 and 4). An IC50 CoMSIA model constructed from the combination of steric (S), electrostatic (E), hydrophobic (H) and hydrogen acceptor (A) fields48 gave the highest q2 (0.646), whereas an MIC90 CoMSIA model including steric, electrostatic, hydrophobic and hydrogen donor (D) fields48 showed the highest q2 (0.639). Therefore, these models were selected for graphical interpretation of IC50 and MIC90 CoMSIA contour maps. In order to assess the predictive abilities of IC50 and MIC90 CoMSIA models, IC50 and MIC90 values of the test set were predicted. Both IC50 and MIC90 CoMSIA models showed good ability to predict IC50 and MIC90 values of the test set data as shown in Fig. 9. In case of IC50 and MIC90 CoMFA models, they had poor predictive ability with q2 values of 0.464 and 0.432, respectively. Accordingly, these CoMFA models were not used further in this work.
Table 3 Statistical results of IC50 CoMFA and CoMSIA modelsa
| Models |
Statistical parameters |
Fraction |
| q2 |
r2 |
s |
SEE |
N |
F |
| Bold values indicate the best CoMSIA model. N optimum number of components; s standard error of prediction; SEE standard error of estimate; F F-test value; S steric field; E electrostatic field; H hydrophobic field; D hydrogen donor field; A hydrogen acceptor field. |
| CoMFA |
| S/E |
0.464 |
0.996 |
0.392 |
0.035 |
6 |
909.618 |
60.3/39.7 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| CoMSIA |
| S/E |
0.084 |
0.977 |
0.512 |
0.081 |
6 |
162.845 |
32.1/67.9 |
| S/H |
0.465 |
0.950 |
0.383 |
0.118 |
5 |
90.431 |
29.1/70.9 |
| S/D |
0.624 |
0.923 |
0.321 |
0.145 |
5 |
57.579 |
54.3/45.7 |
| S/A |
0.146 |
0.970 |
0.495 |
0.093 |
6 |
123.724 |
39.7/60.3 |
| S/E/H |
0.260 |
0.981 |
0.460 |
0.074 |
6 |
194.704 |
16.6/44.5/38.9 |
| S/E/D |
0.592 |
0.980 |
0.342 |
0.076 |
6 |
185.576 |
21.0/53.7/25.3 |
| S/E/A |
0.281 |
0.975 |
0.454 |
0.085 |
6 |
149.701 |
22.5/42.8/34.7 |
| S/E/H/D |
0.646 |
0.990 |
0.318 |
0.055 |
6 |
363.962 |
13.1/35.8/28.5/22.6 |
| S/E/H/A |
0.336 |
0.983 |
0.436 |
0.070 |
6 |
222.520 |
12.3/31.5/29.4/26.8 |
| S/E/H/D/A |
0.610 |
0.991 |
0.334 |
0.050 |
6 |
437.341 |
10.0/25.4/22.6/20.7/21.4 |
Table 4 Statistical results of MIC90 CoMFA and CoMSIA modelsa
| Models |
Statistical parameters |
Fraction |
| q2 |
r2 |
s |
SEE |
N |
F |
| Bold values indicate the best CoMSIA model. N optimum number of components; s standard error of prediction; SEE standard error of estimate; F F-test value; S steric field; E electrostatic field; H hydrophobic field; D hydrogen donor field; A hydrogen acceptor field. |
| CoMFA |
| S/E |
0.432 |
0.853 |
0.442 |
0.225 |
2 |
78.451 |
53.2/46.8 |
|
| CoMSIA |
| S/E |
0.456 |
0.949 |
0.469 |
0.143 |
6 |
71.455 |
25.1/74.9 |
| S/H |
0.459 |
0.780 |
0.432 |
0.275 |
2 |
47.970 |
34.4/65.6 |
| S/D |
0.261 |
0.732 |
0.514 |
0.310 |
3 |
23.642 |
52.7/47.3 |
| S/A |
0.602 |
0.978 |
0.401 |
0.093 |
6 |
174.060 |
46.3/53.7 |
| S/E/H |
0.477 |
0.961 |
0.460 |
0.126 |
6 |
93.558 |
13.8/52.8/33.4 |
| S/E/D |
0.210 |
0.912 |
0.553 |
0.184 |
5 |
49.990 |
17.7/64.4/18.0 |
| S/E/A |
0.550 |
0.955 |
0.426 |
0.134 |
6 |
82.091 |
19.9/48.1/32.0 |
| S/E/H/D |
0.415 |
0.938 |
0.476 |
0.155 |
5 |
72.712 |
10.9/45.8/29.3/13.9 |
| S/E/H/A |
0.639 |
0.973 |
0.382 |
0.105 |
6 |
136.014 |
12.5/35.6/42.2/27.7 |
| S/E/H/D/A |
0.494 |
0.961 |
0.442 |
0.123 |
5 |
118.951 |
9.3/33.4/22.8/10.4/24.2 |
 |
| | Fig. 9 The plot of experimental and predicted activities of the training and test data sets derived from IC50 (a) and MIC90 (b) CoMSIA models. | |
3.8 CoMSIA contour maps
To reveal the importance of molecular descriptor fields in both IC50 and MIC90 values of InhA inhibitors, CoMSIA contour maps were established. Compound 22 presented the best MIC value. Graphical interpretation of its IC50 and MIC90 CoMSIA contour maps was done. Interpretation of its IC50 and MIC90 CoMSIA contour maps revealed structural requirements in terms of steric, electrostatic, hydrophobic and hydrogen donor and acceptor fields for IC50 and MIC90 values of InhA inhibitors.
3.9 Steric requirements for IC50 and MIC90 values
Fig. 10 shows the CoMSIA steric contour maps obtained from selected IC50 and MIC90 CoMSIA models. These contours highlight the steric requirements for IC50 and MIC99 values of benzofuran pyrrolidine pyrazole derivatives. Both IC50 and MIC90 CoMSIA models show a green contour at the R3 substituent. These results indicated that a bulky R3 substituent is favourable for both IC50 and MIC90 values. Accordingly, an ethyl group is more preferred for the steric requirement of the R3 substituent than a methyl group. This is consistent with the MD simulations since an ethyl group can form more interactions with InhA. At the R2 position, IC50 and MIC90 CoMSIA models present a large yellow contour. However, IC50 CoMSIA model shows a favorable green steric contour at the terminal of the R2 substituent (Fig. 10a). Based on MD simulations results, the R2 substituent had weak interaction with the InhA pocket leading to less influence on the IC50 value. Therefore, the steric requirement of R2 substituent should be based on the MIC90 CoMSIA steric contour that presented only a yellow contour near this substituent (Fig. 10b).
 |
| | Fig. 10 Steric contour maps of IC50 (a) and MIC90 (b) CoMSIA models in combination with compound 22. | |
3.10 Electrostatic requirements for IC50 and MIC90 values
Electrostatic requirements for IC50 and MIC90 values of benzofuran pyrrolidine pyrazole derivatives are visualized in Fig. 11. Both IC50 and MIC90 CoMSIA contours show only an electrostatic requirement at the R2 substituent. The IC50 CoMSIA shows a red contour at the ester moiety of R2 substituent, whereas MIC90 CoMSIA presents a blue contour at this position. These results show different electrostatic requirements for IC50 and MIC90 values of benzofuran pyrrolidin pyrazole derivatives. However, the R2 substituent has weak influence on the IC50 value. Therefore, the electrostatic requirement of R2 substituent for MIC90 values should take more priority.
 |
| | Fig. 11 Electrostatic contour maps of IC50 (a) and MIC90 (b) CoMSIA models in combination with compound 22. | |
3.11 Hydrophobic requirements for IC50 and MIC90 values
Both IC50 and MIC90 CoMSIA contours show a purple contour at the R3 substituent of compound 22 (Fig. 12). This shows that the hydrophobic requirements of the R3 substituent for both IC50 and MIC values were similar. The R3 substituent was either a methyl or ethyl group. As seen in Fig. 12, the terminal of ethyl group was buried in a purple R3 contour. Therefore, the ethyl group was preferable for the hydrophobic requirement of the substituent. IC50 and MIC90 values of compound 2 with the methyl group at the R3 substituent were weaker than those of compound 22 containing an ethyl group. At the R2 substituent, both IC50 and MIC90 CoMSIA contours display a purple contour at this position (Fig. 12). Therefore, the presence of a hydrophobic substituent at this purple region should enhance both IC50 and MIC90 values. The grey contour located at the carbonyl moiety of the R2 substituent in both IC50 and MIC90 CoMSIA contours indicated that this moiety is important for both IC50 and MIC90 values. Another important hydrophobic contour is located at the R4 substituent. The MIC90 CoMSIA shows a purple region near the R4 substituent (Fig. 12b), but this contour disappeared in the IC50 CoMSIA contour (Fig. 12a). Therefore, a hydrophobic moiety could be presented at purple region to enhance the MIC90 value without a negative contribution to the IC50 value.
 |
| | Fig. 12 Hydrophobic contour maps of IC50 (a) and MIC90 (b) CoMSIA models in combination with compound 22. | |
3.12 Hydrogen donor and acceptor requirements for IC50 and MIC90 values
The hydrogen donor field was included in the selected IC50 CoMSIA model, but this molecular descriptor was instead changed to a hydrogen acceptor field in the selected MIC90 CoMSIA model (Fig. 13). The IC50 CoMSIA model did not show any hydrogen donor contour near any R substituents. However, this model showed a favourable hydrogen donor contour at the amide moiety of the core structure. The amide moiety appears to impact the IC50 value. Consistent with the MD simulations results, this moiety can form hydrogen bonds with Leu197. The MIC90 CoMSIA model shows a favourable hydrogen acceptor contour at the carbonyl moiety of R2 substituent, indicating that this moiety is essential to a good MIC90 value.
 |
| | Fig. 13 Hydrogen donor contour of IC50 CoMSIA model (a) and hydrogen acceptor contour MIC90 CoMSIA model (b) in combination with compound 22. | |
3.13 The structural concept for good IC50 and MIC90 correlation
Based on the MD simulations results, the core structure of benzofuran pyrrolidine pyrazole derivatives is of key importance for binding in the InhA pocket. Therefore, this fragment is crucial for favorable IC50 values. Among all R substituents, the R2 substituent has the least interaction with the InhA pocket because it protrudes from the pocket. Modifications of the R2 substituent did not significantly change IC50 values, but rather produced a tenfold increase in MIC90 values (compounds 22 and 23). Accordingly, the R2 substituent is a key group that can be used to adjust the MIC90 value without negative contribution to the IC50 value. Based on the results obtained from our MD simulations and CoMSIA studies, the structural concept to correctly balance IC50 and MIC90 values of benzofuran pyrrolidin pyrazole derivatives is summarized in Fig. 14. New compounds designed based on this concept should show better IC50 and MIC90 values.
 |
| | Fig. 14 The structural concept for good IC50 and MIC90 correlation summarized from MD simulations and CoMSIA results. Red and black letters indicate the results obtained from MD simulations and CoMSIA results, respectively. | |
4. Conclusion
The combination of MD simulations and graphical interpretation of IC50 and MIC90 CoMSIA models highlight the structural concept to correctly balance IC50 and MIC90 values of benzofuran pyrrolidin pyrazole derivatives. The core structure of template compound is crucial to attaining favorable IC50 values, whereas the R2 substituent is a key group to enhance MIC90 values without negative effects on IC50 values. Modifications of R substituents following the structural concept suggested here should allow design of novel InhA inhibitors with better potency against both the InhA enzyme and intact M. tuberculosis cells.
Acknowledgements
This research was supported by the Thailand Research Fund (DBG5680003, MRG5680169) the National Research Council of Thailand and Higher Education Research Promotion. The financial support from Royal Golden Jubilee Ph.D. Program (PHD/0004/2554) to P. Kamsri is gratefully acknowledged. Faculty of Science, Ubon Ratchathani University, Kasetsart University and NECTEC are gratefully acknowledged for supporting this research.
References
- Global Tuberculosis Report, World Health Organization, Geneva, 2014 Search PubMed
 .
. - M. C. Becerra, J. Bayona, J. Freeman, P. E. Farmer and J. Y. Kim, International Journal of Tuberculosis and Lung Disease, 2000, 4, 387–394 CAS .
- C. Dye, M. A. Espinal, C. J. Watt, C. Mbiaga and B. G. Williams, J. Infect. Dis., 2002, 185, 1197–1202 CrossRef PubMed .
- M. A. Espinal, Tuberculosis, 2003, 83, 44–51 CrossRef .
- A. Wright, M. Zignol, A. Van Deun, D. Falzon, S. R. Gerdes, K. Feldman, S. Hoffner, F. Drobniewski, L. Barrera, D. van Soolingen, F. Boulabhal, C. N. Paramasivan, K. M. Kam, S. Mitarai, P. Nunn and M. Raviglione, Lancet, 2009, 373, 1861–1873 CrossRef .
- C. Y. Chiang, R. Centis and G. B. Migliori, Respirology, 2010, 15, 413–432 CrossRef PubMed .
- Multidrug and Extensively Drug-Resistant TB (M/XDR-TB):2010 Global Report on Surveillance and Response (WHO/HTM/TB/2010.3), World Health Organization, Geneva, 2010 Search PubMed .
- N. S. Shah, A. Wright, G. H. Bai, L. Barrera, F. Boulahbal, N. Martin-Casabona, F. Drobniewski, C. Gilpin, M. Havelkova, R. Lepe, R. Lumb, B. Metchock, F. Portaels, M. F. Rodrigues, S. RüschGerdes, A. Van Deun, V. Vincent, K. Laserson, C. Wells and J. P. Cegielski, Emerging Infect. Dis., 2007, 13, 380–387 CrossRef CAS PubMed .
- M. Berry and O. M. Kon, Eur. Respir. Rev., 2009, 18, 195–197 CrossRef CAS PubMed .
- A. A. Velayati, P. Farnia, M. R. Masjedi, T. A. Ibrahim, P. Tabarsi, R. Z. Haroun, H. O. Kuan, J. Ghanavi and M. Varahram, Eur. Respir. J., 2009, 34, 1202–1203 CrossRef CAS PubMed .
- Z. F. Udwadia, R. A. Amale, K. K. Ajbani and C. Rodrigues, Clin. Infect. Dis., 2012, 54, 579–581 CrossRef PubMed .
- A. Quemard, J. C. Sacchettini, A. Dessen, C. Vilcheze, R. Bittman, W. R. Jacobs and J. S. Blanchard, Biochemistry, 1995, 34, 8235–8241 CrossRef CAS .
- C. Vilcheze, H. R. Morbidoni, T. R. Weisbrod, H. Iwamoto, M. Kuo, J. C. Sacchettini and W. R. Jacobs Jr, J. Bacteriol., 2000, 182, 4059–4067 CrossRef CAS .
- D. A. Rozwarski, C. Vilchèze, M. Sugantino, R. Bittman and J. C. Sacchettini, J. Biol. Chem., 1999, 274, 15582–15589 CrossRef CAS PubMed .
- D. A. Rozwarski, G. A. Grant, D. H. Barton, W. R. Jacobs Jr and J. C. Sacchettini, Science, 1998, 279, 98–102 CrossRef CAS .
- C. Vilcheze, F. Wang, M. Arai, M. H. Hazbon, R. Colangeli, L. Kremer, T. R. Weisbrod, D. Alland, J. C. Sacchettini and W. R. Jacobs Jr, Nat. Med., 2006, 12, 1027–1029 CrossRef CAS PubMed .
- A. Dessen, A. Quemard, J. S. Blanchard, W. R. Jacobs Jr and J. C. Sacchettini, Science, 1995, 267, 1638–1641 CrossRef CAS .
- K. Johnsson and P. G. Schultz, J. Am. Chem. Soc., 1994, 116, 7425–7426 CrossRef CAS .
- B. Lei, C. J. Wei and S. C. Tu, J. Biol. Chem., 2000, 275, 2520–2526 CrossRef CAS PubMed .
- K. Johnsson, D. S. King and P. G. Schultz, J. Am. Chem. Soc., 1995, 117, 5009–5010 CrossRef CAS .
- Y. Zhang, B. Heym, B. Allen, D. Young and S. Cole, Nature, 1992, 358, 591–593 CrossRef CAS PubMed .
- A. Quemard, A. Dessen, M. Sugantino, W. R. Jacobs Jr, J. C. Sacchetini and J. S. Blanchard, J. Am. Chem. Soc., 1996, 118, 1561–1562 CrossRef CAS .
- A. Banerjee, E. Dubnau, A. Quemard, V. Balasubramanian, K. S. Um, T. Wilson, D. Collins, G. de Lisle and W. R. Jacobs Jr, Science, 1994, 263, 227–230 CAS .
- L. Encinas, H. O'Keefe, M. Neu, M. J. Remuiñán, A. M. Patel, A. Guardia, C. P. Davie, N. Pérez-Macías, H. Yang, M. A. Convery, J. A. Messer, E. Pérez-Herrán, P. A. Centrella, D. Alvarez-Gómez, M. A. Clark, S. Huss, G. K. O'Donovan, F. Ortega-Muro, W. McDowell, P. Castañeda, C. C. Arico-Muendel, S. Pajk, J. Rullás, I. Angulo-Barturen, E. Alvarez-Ruíz, A. Mendoza-Losan, L. Ballell Pages, J. Castro-Pichel and G. Evindar, J. Med. Chem., 2014, 57, 1276–1288 CrossRef CAS PubMed .
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. Millam, M. Klene, J. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. Martin, K. Morokuma, V. Zakrzewski, G. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. Daniels, O. Farkas, J. Foresman, J. Ortiz, J. Cioslowski and D. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed .
- G. Jones, P. Willett and R. C. Glen, J. Mol. Biol., 1995, 245, 43–53 CrossRef CAS .
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed .
- J. W. Nissink, C. Murray, M. Hartshorn, M. L. Verdonk, J. C. Cole and R. Taylor, Proteins, 2002, 49, 457–471 CrossRef CAS PubMed .
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed .
- M. L. Verdonk, G. Chessari, J. C. Cole, M. J. Hartshorn, C. W. Murray, J. W. Nissink, R. D. Taylor and R. Taylor, J. Med. Chem., 2005, 48, 6504–6515 CrossRef CAS PubMed .
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco Search PubMed .
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. A. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed .
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed .
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed .
- P. Cieplak, W. D. Cornell, C. Bayly and P. A. Kollman, J. Comput. Chem., 1995, 16, 1357–1377 CrossRef CAS PubMed .
- W. D. Cornell, P. Cieplak, C. I. Bayly and P. A. Kollmann, J. Am. Chem. Soc., 1993, 115, 9620–9631 CrossRef CAS .
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS .
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS .
- M. W. Mahoney and W. L. Jorgensen, J. Chem. Phys., 2000, 112, 8910–8922 CrossRef CAS PubMed .
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed .
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS .
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed .
- J. Wang, T. Hou and X. Xu, Curr. Comput.-Aided Drug Des., 2006, 2, 95–103 CrossRef .
- J. Wang, P. Morin, W. Wang and P. A. Kollman, J. Am. Chem. Soc., 2001, 123, 5221–5230 CrossRef CAS PubMed .
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed .
- M. Kaledin, A. Brown, A. L. Kaledin and J. M. Bowman, J. Chem. Phys., 2004, 121, 5646–5653 CrossRef CAS PubMed .
- R. D. Cramer III, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef PubMed .
- G. Klebe, U. Abraham and T. Mietzner, J. Med. Chem., 1994, 37, 4130–4146 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08103c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.