
Exploitation of a new Schiff-base ligand for boric acid fluorescent sensing in aqueous medium with bio-imaging studies in a living plant system†
Ambarish
Ray  *b
and
*b
and
*b
and
Abstract
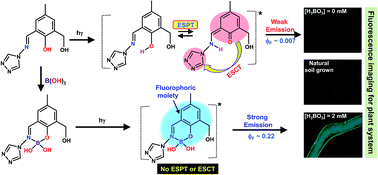
A hydrolysis resistant Schiff-base ligand (L1) consisting of an aromatic aldimine moiety appended to a triazole precursor along with phenolic–OH has been synthesized which acts as a fluorescence probe and exhibits rare selective sensing ability for boric acid (BA) in aqueous buffer medium at pH 7.3 by forming a 1 : 1 chelate complex. Reasons behind the complexation and subsequent fluorescence enhancement are well established using experimental as well as theoretical evidence. Bio-imaging studies are done using Arabidopsis Thaliana as the model plant system.
 Please wait while we load your content...
Please wait while we load your content...

