
Protein–calcium phosphate nanocomposites: benchmarking protein loading via physical and chemical modifications against co-precipitation†
Vinayaraj Ozhukil Kollath‡
*ab,
Steven Mullensa,
Jan Luytenc,
Karl Traina§
d and
Rudi Cloots*b
aDepartment of Chemistry, University of Liège, B6a, Allée de la chimie 3, Liège 4000, Belgium. E-mail: rcloots@ulg.ac.be; Fax: +32 4366 3413
bSustainable Materials Management, Flemish Institute for Technological Research (VITO), Boeretang 200, Mol 2400, Belgium. E-mail: steven.mullens@vito.be; Fax: +32 14 32 11 86
cDepartment of Metallurgy and Materials Engineering, Katholieke Universiteit Leuven, 3001 Heverlee, Belgium
dAPTIS, University of Liège, B5a, Allée de la chimie 3, Liège 4000, Belgium
First published on 18th June 2015
Abstract
The low protein loading capacity of commercially available calcium phosphate (CaP) is a major impediment in effectively using this inorganic material as a protein carrier despite its recognized biocompatibility. In this study, nanocomposites of CaP and BSA were prepared by carefully designed precipitation methods in aqueous media. In the first co-precipitation method (CaP–BSA-1), calcium and phosphate precursors were simultaneously added to the protein solution matrix and in the second method (CaP–BSA-2) the protein solution was added after the reaction of the precursors. The crystallinity and phase composition of the resulting powders were determined using an X-ray diffraction technique. Qualitative confirmation of the presence of BSA on the nanocomposites, was obtained using mass spectrometry, ATR-FTIR and XPS. The results from desorption and thermogravimetric measurements indicated that BSA was trapped inside the cavities in the case of CaP–BSA-1 whereas it was mostly surface adsorbed in the case of CaP–BSA-2. The protein loading capacity of these composites was compared with various physical and chemical surface modification strategies used on commercially available calcium phosphate powders. Nanocomposite particulates were found to have about 275% higher protein loading capacity as compared to a commercial CaP powder with the same surface area. Overall, this study benchmarks the different techniques used for protein loading enhancement on inorganic materials.
Introduction
Protein adsorption on inorganic materials is a complex paradigm, which finds applications in various biomedical issues. Several attempts have been made in order to understand the different mechanisms controlling the protein adsorption on various materials.1–5 Natural adsorption of proteins on inorganic materials can effectively be utilized to develop inorganic carrier-protein delivery systems.6 Calcium phosphate (CaP) is highly suitable to use as an inorganic carrier, due to its biocompatibility and resorbability. Surface modifications on commercially available CaP powders have been performed using physical and chemical methods to improve the protein adsorption capacity. These modifications demonstrated the physical parameters as well as the surface chemical groups affecting the loading capacity on β-tricalcium phosphate and hydroxyapatite.7–11A limitation of CaP powder as a carrier material is the particle agglomeration in aqueous suspensions.12 This agglomeration mainly arises from charge neutrality on the particles in an aqueous suspension, which may hinder the possibility to test them in vivo due to size limitations.13,14 Possible solution to overcome the agglomeration problem is to make these particles charged in situ in order to give inherent electrostatic stability in aqueous medium. Bovine serum albumin (BSA) macromolecules are incorporated during the precipitation of the CaP particles, with a view to increase the inherent charge of CaP particles using the negative charge of BSA macromolecules in aqueous solution at neutral pH. It has been reported that proteins and amino acids can control the growth of CaP crystals15–20 and hence influence the size and crystallinity.
This study discusses the potential of co-precipitated CaP–BSA nanocomposite particles to enhance the protein loading and simultaneously increase the stability of carrier-protein suspension. Three different CaP powders (includes control sample) were prepared using co-precipitation routes. The incorporated BSA was characterized both quantitatively and qualitatively. The potential of co-precipitation method has been represented by plotting developed CaP nanocomposites' protein loading capacity with respect to the estimated surface area. This was compared with protein loading on commercially available CaP powders as well as the different surface functionalization methods used on them, viz. physical modification,8 atmospheric pressure plasma modification9 and wet-chemical functionalization.10,11
Experimental
Materials
Commercially available calcium nitrate tetrahydrate [Ca(NO3)2·4H2O; Merck], di-ammonium hydrogen phosphate [(NH4)2·HPO4; Merck], ammonium hydroxide [NH4OH; Acros Organics] and Bradford reagent, BSA [Sigma-Aldrich] were used without further purification. Highly pure water [MilliQ water; Elix, Millipore] was used as solvent for salt solutions. Ammonium hydroxide (25% solution in milliQ water) was used in order to adjust the pH if necessary.Synthesis
Three types of CaP powders were obtained using co-precipitation methods. In the first case, CaP was precipitated within the BSA solution matrix (named as CaP–BSA-1) whereas in the second case the precipitation occurred before the addition of BSA solution (named as CaP–BSA-2). The third type of powder was prepared as a reference where the precipitation occurred in aqueous environment (named as CaP-0). The physico-chemical characteristics of these powders are different due to the various reaction matrices applied in these protocols. A general schematic of synthesis set-up is shown in Fig. 1 and step-by-step details are represented in Scheme S1 (ESI†). Detailed protocols are described as follows: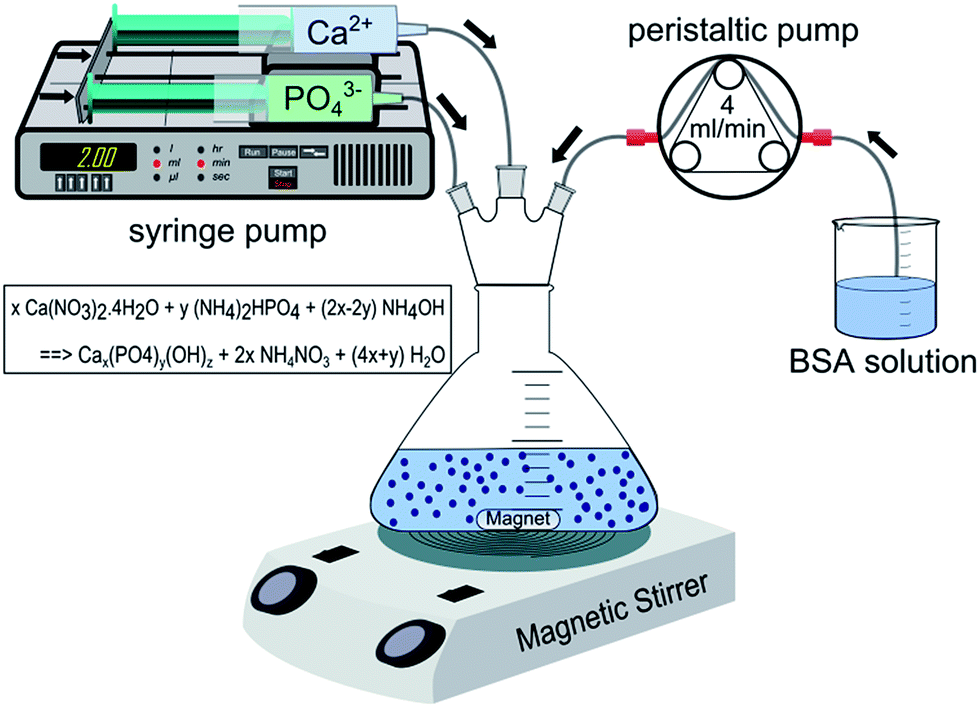 | ||
| Fig. 1 Schematic of the synthesis set-up used in this study. Arrow marks show the direction of flow. The chemical reaction shown excludes the BSA. For x/y = 1.67, a complete reaction should ideally yield a stoichiometric hydroxyapatite. | ||
(2) 40 ml of Ca(NO3)2·4H2O (50 mM; pH 10.5) and (NH4)2HPO4 (30 mM; pH 9) was pumped simultaneously to the conical flask at 2 ml min−1 using a syringe pump (NE1000, ProSense BV). A 50 ml syringe with an inner diameter of 28.4 mm was used. Stirring of the solution mixture was continued, which became a turbid white suspension.
(3) Ageing of the resulted suspension was done by further stirring at 250 rpm (room temperature) for 4 h. No heating was applied to avoid denaturation of BSA above 50 °C [Brandes et al., 2006].
(4) After ageing, the suspension was centrifuged (Centrifuge 5810, Eppendorf AG) at 5000 rpm for 10 min.
(5) Supernatant (supernatant 1) was collected for BSA quantification. The rest of the supernatant was decanted.
(6) 40 ml of milliQ water was added to the centrifuge tube, re-dispersed and centrifuged again at 5000 rpm for 10 min. Supernatant (supernatant 2) was collected for BSA quantification. This step acted as a washing step to remove the excess ions and any loosely bound BSA.
(7) Previous step was repeated one more time (supernatant 3).
8.The resulting paste was dried in a Petri dish at 30 °C for 72 h in an oven with air flow.
(2) 20 ml of BSA solution (20 mg ml−1) was added to the resulting suspension using a peristaltic pump at 4 ml min−1 (speed 9, Flocon 1000, Periflo) immediately after the end of reactants injection.
(3) The resulting turbid white suspension was aged for 18 h while stirring at room temperature.
(4) Washing and drying steps were carried out in the same way as mentioned for CaP–BSA-1.
Several parameters could be varied to control the precipitation reactions, such as concentration of the reactants, flow rate, temperature and precursor mixing speed, pH of precursors, etc. Aqueous solutions of Ca(NO3)2·4H2O (50 mM) and (NH4)2·HPO4 (30 mM) were used as precursors. Adapted from literature,21 the pHs were adjusted to 10.5 and 9 for Ca(NO3)2·4H2O and (NH4)2·HPO4, respectively. Both the pH adjustments were carried out using NH4OH and measured using a pH meter (SevenMulti, Mettler Toledo). The concentrations of the reactants were selected so that the Ca/P ratio = 1.67 (same as hydroxyapatite). The reactions were performed at room temperature (∼23 °C) in order to avoid any denaturation of the BSA from higher temperature synthesis.
Characterisation
The crystalline phases of the resulted powders were detected using X-ray diffraction technique (XRD; X'pert, PANalytical, The Netherlands) with a Cu-Kα X-ray source (λ = 1.54056 Å). Surface morphology and elemental composition of the powders were recorded with a scanning electron microscope equipped with an energy dispersive spectrometer (SEM-EDS; JSM-6340F, Jeol, Japan). Particle size distributions of the powders were measured using laser diffraction technique (LS230, Beckman Coulter, USA). The suspensions used for particle size analysis were ultrasonicated using an ultrasonic horn (13 mm; Vibracell VCX600, Sonics & Materials Inc., U.S.A.). The ultrasonications were 20 s at 50% amplitude and alternating pulse of 1 s. Power of the US set up is 600 W. Due to the low sample amount, Kr-sorption was used to measure the specific surface area (SSA). The Kr-sorption measurement at −186 °C was determined using a Quantachrome Quadrasorb SI automated gas sorption system. Prior to the measurement, the sample CaP-0 was outgassed under vacuum during 48 h at a temperature of 150 °C. In the case of BSA containing powders, the degassing temperature was kept at 40 °C. The Brunauer-Emmet-Teller (BET) method was applied to calculate the apparent specific surface area (SSA). Bradford assay was used to quantify the BSA present in each case. Bradford reagent was mixed with BSA containing solution, according to manufacturer's protocol. Absorbance of the complex formed was measured at 595 nm using a UV-visible spectrometer and the amount of BSA present was calculated from the calibration curve. The amount of BSA co-precipitated was calculated from the difference between the initial concentration and the supernatant concentration. Complementary quantification of the BSA was performed using thermogravimetric analysis (TGA; STA 449C, Netzsch, Germany). TGA measurements were performed by heating pre-weighted samples up to 1000 °C in dry air atmosphere, at a rate of 10 °C min−1. Mass spectra (MS) were obtained online during the TGA measurements. Elemental analysis was performed on BSA incorporated samples using XPS (Thetaprobe, Thermo electron corporation, U.K.). Fourier transform infrared (FTIR) spectra of samples were recorded in attenuated total reflectance (ATR) mode using diamond crystal (Nicolet Nexus, Thermo electron corporation, U.S.A.).Results and discussion
Powder characterisation
The X-ray diffraction patterns are presented in Fig. 2, together with the peak lists of the detected crystalline phases, namely hydroxyapatite (HA; Ca5(PO4)3OH), carbonated hydroxyapatite (CHA; Ca10(PO4)5.65(CO3)0.64(OH)3.452), calcium hydroxide (Ca(OH)2) and brushite (CaPO3(OH)·2H2O). In all these cases, the peaks were broad which indicates the small size of the particles. For this reason, it was difficult to assign only one phase between HA and CHA phases. The presence of CHA has been discussed subsequently. Fig. 2 shows peaks of HA/CHA and calcium hydroxide for the powder CaP-0. Pattern of CaP–BSA-1 powder was very similar to that of CaP-0, but the peak intensities were lesser which could be due to the low amount of sample used. The CaP–BSA-2 powder showed resolved peaks of brushite phase in addition. A complete evaluation of the XRD spectra was not carried out since the focus of this study was to compare the protein loading strategies.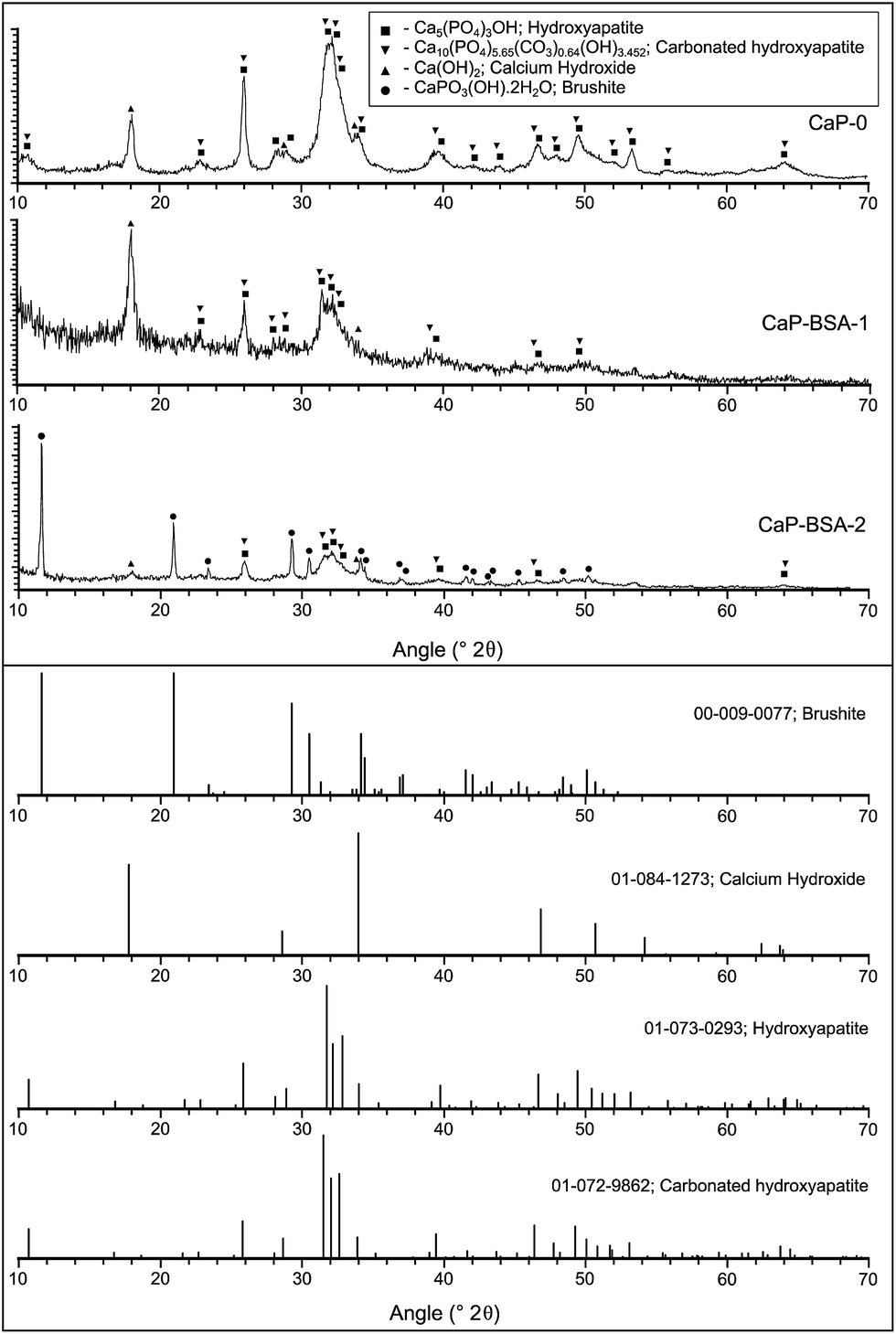 | ||
| Fig. 2 XRD spectra of CaP-0, CaP–BSA-1 and CaP–BSA-2 and corresponding peak lists of the detected phases. | ||
The crystallite sizes of HA/CHA phase were calculated using Scherrer formula (eqn. (1)). According to this formula, the mean crystallite size:
 | (1) |
The mean crystallite sizes calculated were 26.5, 21.9 and 22.5 nm for CaP-0, CaP–BSA-1 and CaP–BSA-2 respectively. These values should be considered as rough estimates, as the peaks were of low intensity which could affect the calculation due to the noise in the pattern. However, this indicates the inhibition tendency of BSA present in the case of CaP–BSA-1 and CaP–BSA-2 crystal growth.
Fig. 3 shows the morphologies of the synthesized powders. The powder CaP-0 showed mainly ellipsoid type particles (Fig. 3a and b) strongly agglomerated to irregular shapes. Both the powders CaP–BSA-1 (Fig. 3c and d) and CaP–BSA-2 (Fig. 3e and f) after washing, consisted of soft agglomerates of flake-like structures of 70–120 nm in length and approximately 50 nm in width. Similar flake-like structures were reported by Hagmeyer et al. for the CaP nanoparticles prepared in presence of tyrosine phosphate and similar amino acids.22
 | ||
| Fig. 3 Scanning electron micrographs of CaP-0 (a and b) CaP–BSA-1 (c and d) and CaP–BSA-2 (e and f). The scale bars are 1 μm (left) and 100 nm (right) in each panel. | ||
Agglomeration was visible in dried forms, a known phenomenon for CaP prepared from aqueous suspensions.23 However the softer agglomerates in case of CaP–BSA-1 and CaP–BSA-2 indicated their lower aggregation in colloidal state and hence higher stability.
Energy dispersive spectra of the synthesized powders are shown in ESI material (Fig. S1†). The spectrum of CaP-0 (Fig. S1a†) showed characteristic peaks of calcium, phosphor, oxygen and carbon. The presence of carbon could be attributed to the carbonated phase present in the powder, as seen in the XRD spectra.
The spectra from CaP co-precipitated with BSA (Fig. S1b and c†) showed peaks of carbon, oxygen, calcium and phosphor in the structure. The carbon peak intensity is much higher for these samples whereas the calcium and phosphor peak intensity was reduced as compared to the spectrum of CaP-0. These details could be attributed to the presence of co-precipitated BSA.
Fig. 4 shows the particle size characterisation results. The particle number distribution CaP–BSA-1 reveals that the major portion of the resulting powder is submicron sized and majority of the particle population is below 200 nm. The number distribution of CaP–BSA-2 is relatively narrow as compared to CaP–BSA-1. Again, it was largely contributed by particles with size less than 200 nm. In both the cases, maxima of number distribution curves are approximately 70 nm. These results confirm the softness of the agglomerates observed by SEM-EDS.
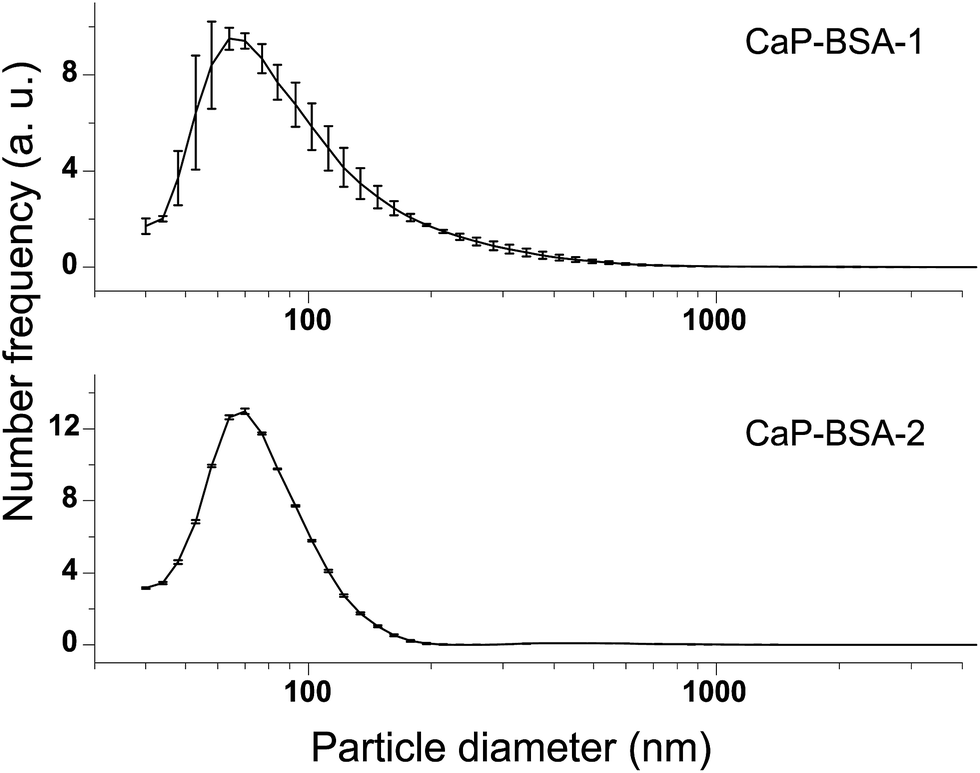 | ||
| Fig. 4 Particle size distribution of CaP–BSA-1 and CaP–BSA-2. | ||
The SSA of prepared powders was measured using Kr-sorption method. The SSA measurement of CaP-0 powder yielded a value of 84 m2 g−1. The Kr-sorption measurements could not be completed on CaP powders co-precipitated with BSA. One possible reason is that the SSA of these powders is less than the lower detection limit of Krypton adsorption at 87 K. The lower limit of the sample cell is 0.2 m2, which corresponds to an SSA < 2 m2 g−1 for 0.1 g powder sample measured. However, this is highly unlikely considering the nano-size of these particles. Thus it can be imagined that these powders are incorporated in a BSA matrix which is not accessible for Kr gas molecules. This reasoning is supported by the successful Kr adsorption on the powder prepared without BSA (CaP-0).
BSA loading
The yield of powders produced was estimated using gravimetric method. After the two washing and re-dispersion in milliQ water (the suspension volume before and after washing were the same), 20 ml of powder suspension was taken in a 250 ml pre-weighed glass beaker. The suspension was weighed and then dried in air at 40 °C during 5 days to remove the water content. The beaker containing dry powder was weighed to calculate the amount of powder as well as the volume of water evaporated. This gave an estimate of powder present per ml of the suspension. The calculated values were 6.99 and 3.86 mg ml−1 for CaP–BSA-1 and CaP–BSA-2, respectively.The amounts of stock BSA solution, available BSA on the powder after each centrifugation and the corresponding pH of the supernatants are shown in Table 1.
| CaP–BSA-1 | CaP–BSA-2 | |||
|---|---|---|---|---|
| BSA remained (mg) | pH | BSA remained (mg) | pH | |
| BSA stock solution | 396.3 | 377.1 | ||
| After first centrifugation | 237.6 | 5.8 | 125.7 | 5.8 |
| After second centrifugation | 220.7 | 6.7 | 115.4 | 6.5 |
| After third centrifugation | 220.7 | 7 | 115.4 | 7 |
From the calculated amount of powders produced and the remaining BSA after the washing step, the amount of BSA co-precipitated was calculated.
BSA co-precipitated per gram of CaP–BSA-1 = 316 mg.
BSA co-precipitated per gram of CaP–BSA-2 = 299 mg.
Complementary to the above quantification, TGA shows the weight loss as a function of temperature from which the amount of incorporated BSA can be calculated. An on-line MS measurement helps to determine the gases released during the TGA. The weight loss curve of the CaP-0 powder (Fig. 5a) shows an initial three stage weight loss between 30 and 200 °C, with DTG maxima at 80, 140 and 195 °C. This corresponds to the loss of loosely and strongly adsorbed water. Further a gradual decrease in the TG curve is observed until 400 °C, which is attributed to the release of lattice water.24 Additionally, the water released during the decomposition of Ca(OH)2 phase (as detected by XRD) of CaP-0 can be attributed to the DTG maximum at 430 °C.25 The final small mass loss with a DTG maximum at 544 °C is attributed to the release of CO2 due to the initial decomposition of phases containing amorphous carbonate. This can be confirmed from the XRD results (Fig. 2), which showed the presence of carbonated phases of HA present in this powder.
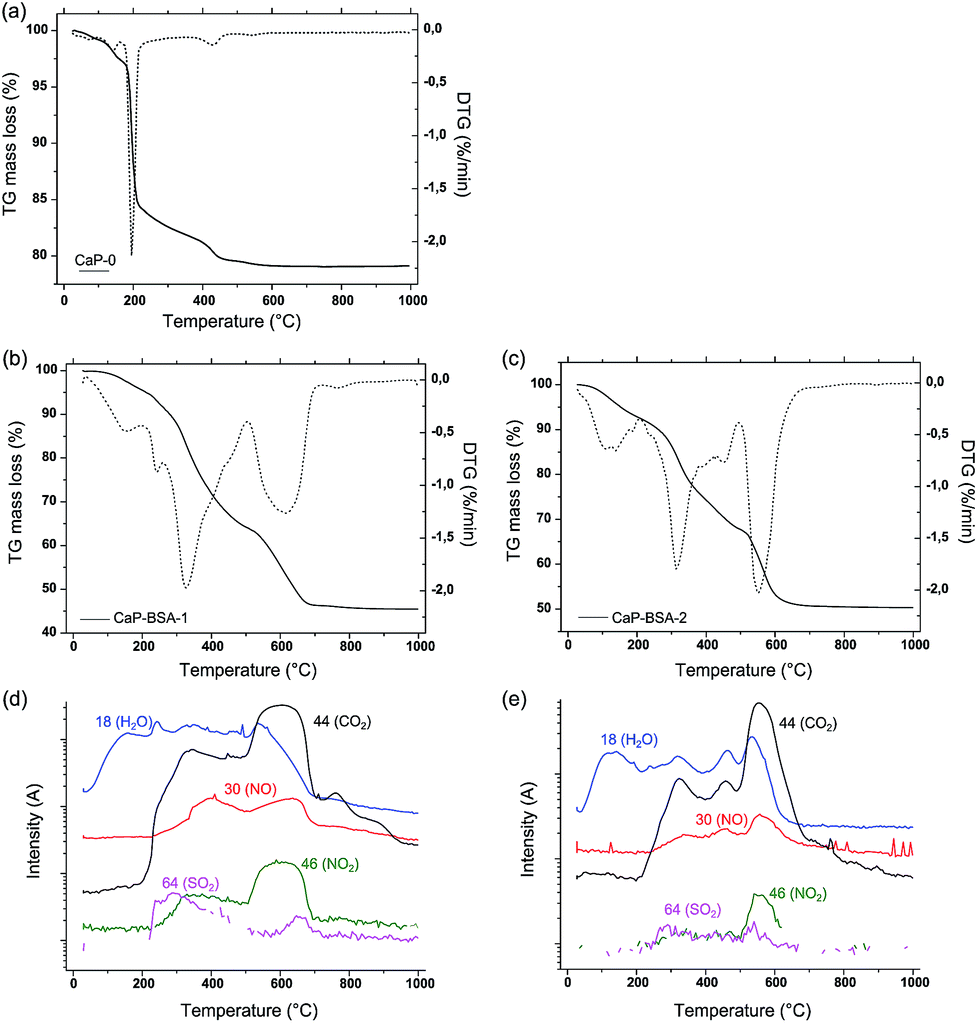 | ||
| Fig. 5 Thermo gravimetric spectra of (a) CaP-0, (b) CaP–BSA-1 and (c) CaP–BSA-2. The solid curves (in a–c) represent the TG axis and dotted curves represent the respective differential spectra. Corresponding mass spectra of (b) and (c) are given as (d) and (e) respectively. | ||
For powders co-precipitated with BSA, desorption of surface water was observed at a DTG maxima approximately around 100–150 °C (Fig. 5b and c). A higher weight loss was detected for both the powders in the temperature range 200–500 °C. There are three DTG maxima visible in this temperature range. Compared to the corresponding MS (Fig. 5d and e), this weight loss can be assigned to the oxidation and thermal decomposition of incorporated BSA with the release of CO2 (m/z = 44), H2O (m/z = 18), NO2 (m/z = 46), NO and/or amines (m/z = 30) and SO2 (m/z = 64), which are overlapped with the decomposition of amorphous phases of the CaP powder. The SO2 release comes from the sulphur content of the BSA which is not observed in the MS of CaP-0. Another major weight loss is observed on both of these powders in the temperature range 500–700 °C. There is a final small percentage weight loss at around 800 °C, in these powders. Only CO2 release is observed in the corresponding MS, which is attributed to the release of CO2 from the decomposition of crystalline carbonated HA.
In general, all the DTG maxima of CaP–BSA-1 are shifted to higher temperatures as compared to that of CaP–BSA-2. One possible reason is the difference in preparation protocols of these powders, which results in different crystallinity of the phases or differences in the microstructural matrix of the samples. In the case of CaP–BSA-1, BSA could be hypothesised as trapped inside the interstitial pores of CaP particulates (Scheme 1) and hence with stronger bond with CaP. On the other hand, the CaP–BSA-2 will have the BSA adsorbed on the outer surface of the CaP since the nucleation takes place before addition of BSA solution. Thus it can be inferred from the TGA spectra that a surface adsorbed BSA (CaP–BSA-2) will have complete combustion at lower temperature as compared to that trapped within the structure of CaP (CaP–BSA-1). The differences in weight loss with respect to CaP-0 were 33.7 and 29.2% for CaP–BSA-1 and CaP–BSA-2, respectively. These values are in good agreement with the photometric quantification of adsorbed BSA using Bradford assay.
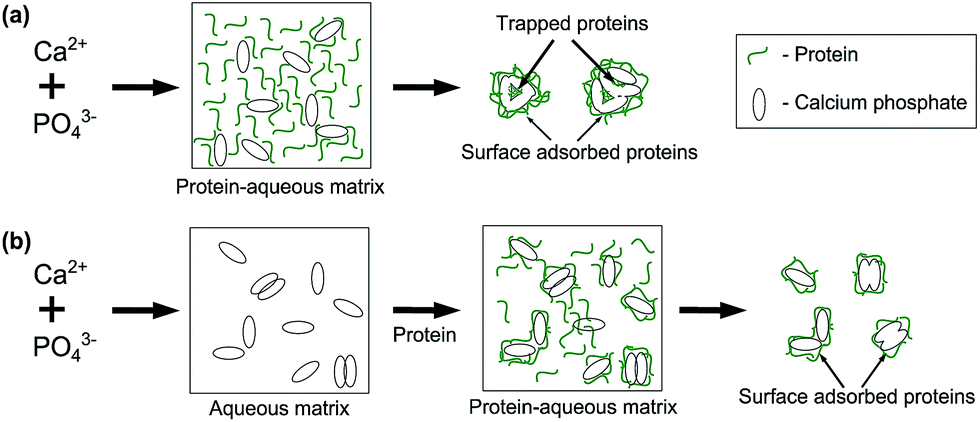 | ||
| Scheme 1 Hypothetical representation of the BSA loading in the case of (a) CaP–BSA-1 and (b) CaP–BSA-2. | ||
From the results discussed above, it can be concluded that the BSA loading to the CaP powder could be increased significantly by the selected synthesis routes. A general comparison of the developed synthesize routes to the adsorption on commercially available CaP powders, is given in the following sub-section.
In order to further evaluate the BSA detected using TGA-MS, XPS analysis was performed on the BSA containing samples. The results (Table 2) verified the presence of BSA from the high atomic percentage of nitrogen detected. The amount of nitrogen detected was higher in the case of CaP–BSA-1, which is in agreement with the TGA and Bradford assay results. Reduction in the atomic percentage of calcium and phosphor is an indication of CaP incorporated inside the BSA matrix in these co-precipitated powders. This observation strengthens the non-adsorption of Kr during SSA measurements.
ATR-FTIR was applied on the synthesized powders for qualitative analyses of both the CaP and BSA present in the co-precipitated powders. Fig. 6 shows the spectra of all three powders. From the XRD results, the presence of carbonated apatite (CHA) was noted. In general, CHA is classified according to the carbonate substitution sites. In type-A CHA, the carbonate ions occupies the OH sites; in type-B CHA, they occupy the PO4 sites and in type-AB they occupy in both sites.26,27 For carbonates, IR bands between 1540 and 1420 cm−1 corresponds to the in-plane asymmetric stretch vibration mode and the band within 880–870 cm−1 corresponds to the out-of-plane bending mode.26 In case of CaP-0, only one weak band is visible at 1457 cm−1 within the in-plane asymmetric stretch region. However the band at 870 cm−1 indicates the presence of type-B CHA present in CaP-0.26,27 In addition, the bands at 3570 and 629 cm−1 are assigned to the OH group. Thus it can be concluded that the CHA phase present is not type-A. However the OH groups are also present in the calcium hydroxide phase recognized in the XRD spectra. Thus in combination with the XRD results, the CHA phase present in CaP-0 can be assumed as type-AB CHA. The band at 1653 cm−1 was assigned to the surface adsorbed water. The IR bands marked between 1200 and 900 cm−1 and between 605 and 475 cm−1 are characteristic phosphate group bands.4,28
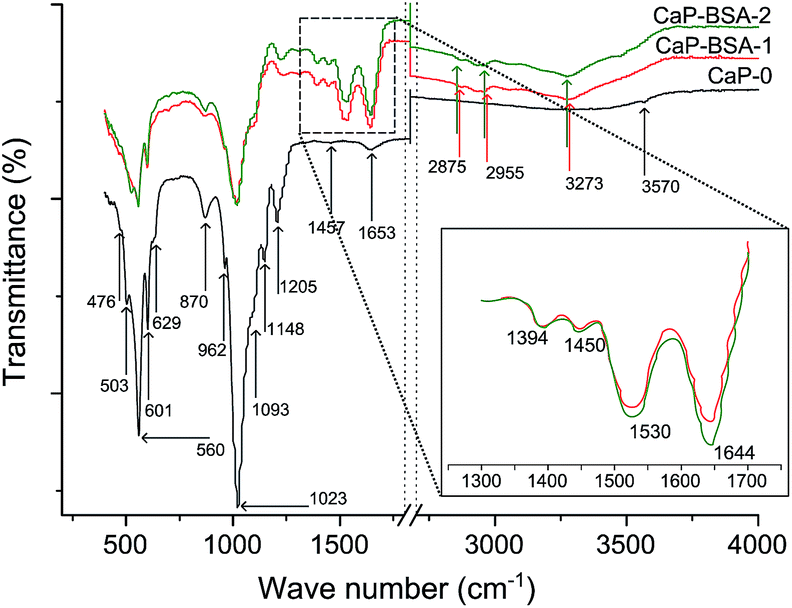 | ||
| Fig. 6 ATR-FTIR spectra of CaP-0 (black), CaP–BSA-1 (red) and CaP–BSA-2 (green). The inset shows the amide band region. Vertical dotted lines are to notify the X-axis break. | ||
For the powder co-precipitated with BSA (Fig. 6 inset), the characteristic bands of BSA was present in the form of amide and carboxyl group vibrations. The bands at 1644, 1530 and 1394 cm−1 corresponds to the amide I, II and III vibrations respectively. The band at 1450 cm−1 was assigned to the carboxyl group vibration and the bands in the region 3000–2800 cm−1 are characteristic of the alkane chains of BSA.8,29
A sedimentation test (visual examination) was done on CaP–BSA-2 to check the stability of the suspension. A part of the freshly prepared suspension after washing was taken in a glass test tube and kept stationary. The liquid column stayed turbid up to 4 h with minimal sedimentation whereas a suspension containing BSA adsorbed on the commercial HA powder sedimented completely within 30 min. This is indicative of the higher stability of co-precipitated CaP–BSA powders.
Results from the desorption measurements (Table 1) and TGA (Fig. 5) indicate that BSA is trapped inside the cavities in the case of CaP–BSA-1 whereas it is mostly surface adsorbed in the case of CaP–BSA-2. Combining the results discussed above and the model reported by Combes and Rey30 on the nucleation and growth process of calcium phosphate in the presence of protein, the nanocomposites prepared in this study can be visualised to have two types of protein loading. This is represented in Scheme 1.
Comparison of protein loading strategies
From the results published in our previous studies and additional experiments performed in this study, an overview of the BSA adsorption with respect to various surface modification routes applied on the commercial CaP powders is shown in Fig. 7 (refer ESI† for properties of commercial powders used).8–10 The powders without any chemical modifications show linear increase with respect to their SSA. This is valid for both β-tricalcium phosphate as well as hydroxyapatite phases. The chemical processing routes, namely the wet-chemical functionalisation as well as the atmospheric pressure (AP) plasma activation showed an increase of up to 66% in BSA surface adsorption. The increased protein adsorption of the HA powder with SSA = 90 m2 g−1 after arginine functionalisation indicated that the wet-chemical functionalisation can be applied independent of the SSA.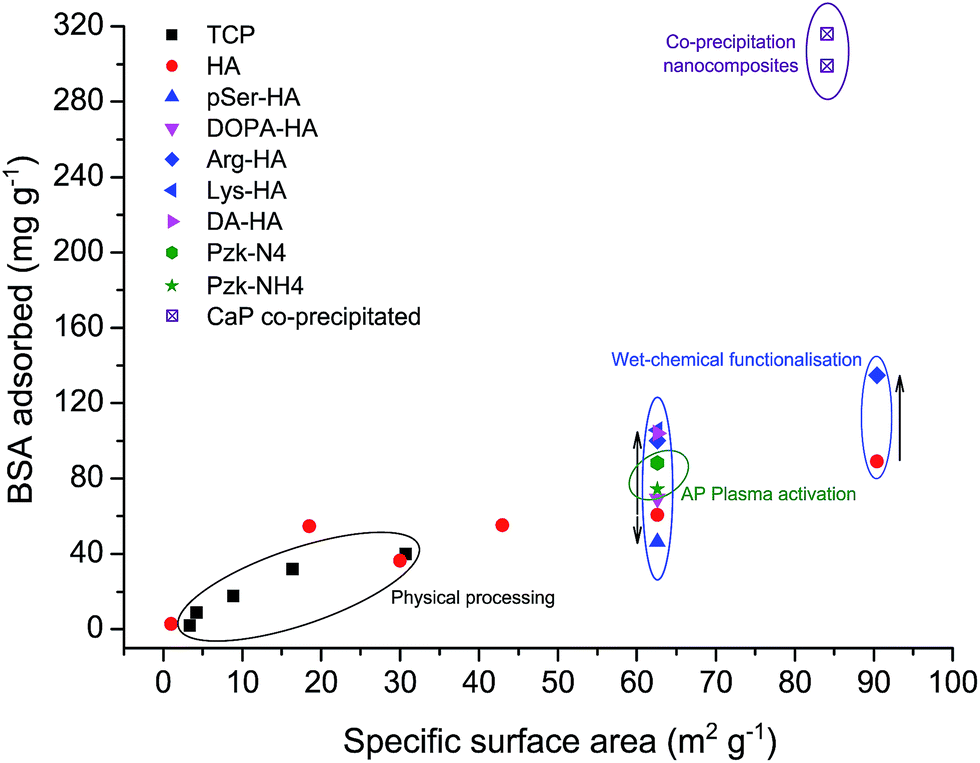 | ||
| Fig. 7 Global overview of the mean amount of BSA adsorbed per gram of different CaP powders, with respect to their SSA; and routes used. Closed square: β-tricalcium phosphate (TCP); circle: hydroxyapatite (HA); up-triangle: phosphoserine-HA; down-triangle: 3,4-dihydroxyphenylalanine-HA; diamond: arginine-HA; left-triangle: lysine-HA; right-triangle: dopamine-HA; hexagon: N2 atmospheric pressure plasma activated HA; star: N2 + H2 atmospheric pressure plasma activated HA; crossed square: nanocomposites in this work. Initial BSA concentrations are 4 mg ml−1 for co-precipitation method, and 10 mg ml−1 for the rest [refer ESI,†8–11]. | ||
A global comparison of all the commercial powders used in this and earlier studies, to the synthesized CaP nanocomposites is marked in Fig. 7. The initial BSA concentration used was 10 mg ml−1 for commercial powders, whereas 4 mg ml−1 was present during the co-precipitation. The adsorption capacity reached up to 300 mg g−1 for co-precipitated powders. The amounts of BSA incorporated with the nano CaP–BSA composites were approximately 275% higher than that on a commercial powder of SSA = 84 m2 g−1 (comparative estimation). The linear relation between protein adsorption and the SSA was no more applicable to the co-precipitated powders. A possible explanation can be that the protein could interact with more active surface of CaP nanocomposites. Thus the co-precipitation method proves to be more advantageous over the physical and chemical processing methods, if the phase purity is not an issue. However, various optimisation procedures for the phase purity, crystallinity and dissolution are recommended in line with their effect in the biological systems.
Conclusions
This study explored the potential use of CaP–BSA co-precipitation method to prepare CaP powder with high protein loading capacity. Two types of co-precipitated CaP–BSA composites were successfully achieved by the in situ aqueous non-thermal synthesis route. These composite powders showed different phase compositions but similar particle morphology. The BSA is either incorporated (CaP–BSA-1) or adsorbed (CaP–BSA-2) during the co-precipitation. These powders could be considered as nanocomposites, as the major population of the number distributions were within 100 nm. A high loading capacity was observed in both the powders. Also the stability of these nanocomposite suspensions improved considerably over similar concentration suspensions made from commercial powders. The nanocomposites prepared in this study via co-precipitation method can be considered as an adaptable example for variety of protein–inorganic material systems which require higher protein loading and better stability as an aqueous formulation.Acknowledgements
VOK gratefully acknowledges the financial support from University of Liège and VITO. The authors thank S.J. Herregods, V. Meynen (Univ. Antwerp, Belgium) for Kr-sorption SSA measurements and interpretation; and the technical support from R. Kemps, M. Mertens, H. Chen, A.M. De Wilde and I. Thijs (VITO).References
- K. Nakanishi, T. Sakiyama and K. Imamura, J. Biosci. Bioeng., 2001, 91, 233–244 CrossRef CAS.
- K. Wang, C. Zhou, Y. Hong and X. Zhang, Interface Focus, 2012, 2, 259–277 CrossRef PubMed.
- K. Rezwan, L. P. Meier, M. Rezwan, J. Vörös, M. Textor and L. J. Gauckler, Langmuir, 2004, 20, 10055–10061 CrossRef CAS PubMed.
- S. Dasgupta, A. Bandyopadhyay and S. Bose, Acta Biomater., 2009, 5, 3112–3121 CrossRef CAS PubMed.
- M. Alkan, Ö. Demirbaş, M. Doğan and O. Arslan, Microporous Mesoporous Mater., 2006, 96, 331–340 CrossRef CAS PubMed.
- Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS PubMed.
- W. Lee, C. Loo, K. L. Van, A. V. Zavgorodniy and R. Rohanizadeh, J. R. Soc., Interface, 2012, 9, 918–927 CrossRef CAS PubMed.
- V. Ozhukil Kollath, B. G. De Geest, S. Mullens, S. De Koker, J. Luyten, R. Persoons, K. Traina, J. P. Remon and R. Cloots, Adv. Eng. Mater., 2013, 15, 295–301 CrossRef CAS PubMed.
- V. Ozhukil Kollath, S. Put, S. Mullens, A. Vanhulsel, J. Luyten, K. Traina and R. Cloots, Plasma Processes Polym., 2015, 12, 594–601, DOI:10.1002/ppap.201400092.
- V. Ozhukil Kollath, F. Van Den Broeck, K. Fehér, J. C. Martins, J. Luyten, K. Traina, S. Mullens and R. Cloots, Chem.–Eur. J., 2015 DOI:10.1002/chem.201500223.
- V. Ozhukil Kollath, S. Mullens, J. Luyten, K. Traina and R. Cloots, Materials Technology: Advanced Performance Materials, 2015 DOI:10.1179/1753555715Y.0000000048.
- V. Uskoković and D. P. Uskoković, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef PubMed.
- J. A. Cohen, T. T. Beaudette, W. W. Tseng, E. M. Bachelder, I. Mende, E. G. Engleman and J. M. J. Fréchet, Bioconjugate Chem., 2009, 20, 111–119 CrossRef CAS PubMed.
- C. V. Harding and R. Song, J. Immunol., 1994, 153, 4925–4933 CAS.
- S. V. Dorozhkin and E. I. Dorozhkina, Colloids Surf., A, 2003, 215, 191–199 CrossRef CAS.
- S. Koutsopoulos and E. Dalas, Langmuir, 2001, 17, 1074–1079 CrossRef CAS.
- S. Koutsopoulos and E. Dalas, J. Cryst. Growth, 2000, 216, 443–449 CrossRef CAS.
- S. Koutsopoulos and E. Dalas, J. Colloid Interface Sci., 2000, 231, 207–212 CrossRef CAS PubMed.
- R. Gonzalez-mcquire, J. Chane-ching, E. Vignaud, A. Lebugle and S. Mann, J. Mater. Chem., 2004, 14, 2277–2281 RSC.
- K. S. Jack, T. G. Vizcarra and M. Trau, Langmuir, 2007, 23, 12233–12242 CrossRef CAS PubMed.
- E. Hayek, H. Newesely and M. L. Rumpel, in Inorganic Syntheses, ed. J. Kleinberg, John Wiley & Sons, Inc., Hoboken, NJ, USA, 7th edn., 1963, pp. 63–65 Search PubMed.
- D. Hagmeyer, K. Ganesan, J. Ruesing, D. Schunk, C. Mayer, A. Dey, N. A. J. M. Sommerdijk and M. Epple, J. Mater. Chem., 2011, 21, 9219–9223 RSC.
- V. Sokolova, A. Kovtun, O. Prymak, W. Meyer-Zaika, E. A. Kubareva, E. A. Romanova, T. S. Oretskaya, R. Heumann and M. Epple, J. Mater. Chem., 2007, 17, 721 RSC.
- L. Sun, L. C. Chow, S. A. Frukhtbeyn and J. E. Bonevich, J. Res. Natl. Inst. Stand. Technol., 2010, 115, 243–255 CrossRef CAS.
- G. Villain, M. Thiery and G. Platret, Cem. Concr. Res., 2007, 37, 1182–1192 CrossRef CAS PubMed.
- T. Tonegawa, T. Ikoma, T. Yoshioka, N. Hanagata and J. Tanaka, J. Mater. Sci., 2010, 45, 2419–2426 CrossRef CAS.
- M. E. Fleet and X. Liu, Biomaterials, 2007, 28, 916–926 CrossRef CAS PubMed.
- I. Rehman and W. Bonfield, J. Mater. Sci.: Mater. Med., 1997, 8, 1–4 CrossRef CAS.
- S. Dasgupta, S. S. Banerjee, A. Bandyopadhyay and S. Bose, Langmuir, 2010, 26, 4958–4964 CrossRef CAS PubMed.
- C. Combes and C. Rey, Biomaterials, 2002, 23, 2817–2823 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Schematics of nanocomposite synthesis, EDS spectra, SSA and SEM of commercial CaP powders used. See DOI: 10.1039/c5ra08060f |
| ‡ Current address: Department of Physics, UNESP—Univ. Estadual Paulista, Bauru, SP, 17033-360, Brazil. |
| § Current address: Galephar MF, 6900 Marche en Famenne, Belgium. |
| This journal is © The Royal Society of Chemistry 2015 |