
Self-assembly, photoresponsive behavior and transport potential of azobenzene grafted dendronized polymeric amphiphiles†
Abstract
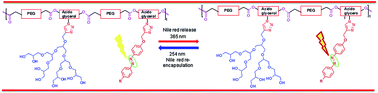
Photoresponsive polymeric amphiphiles were developed by first synthesizing the polyester chain via Novozym 435 catalyzed step growth, condensation polymerization of poly[ethylene glycol bis(carboxymethyl) ether]diethylester and 2-azidopropan-1,3-diol followed by grafting with 4′-butyl-4-propargyloxy(azobenzene) and [G2.0] polyglycerol dendron by using a ‘Click chemistry’ approach. The resulting polymers were observed to form supramolecular micellar aggregates in aqueous solution. The critical aggregation concentration (CAC) was determined via fluorescence measurements and using ‘Nile red’ as a probe. The nano-structures formed in the aqueous solution were characterized by dynamic light scattering (DLS) and cryo-TEM measurements. The encapsulation potential of polymeric amphiphiles for Nile red and curcumin as well as their release via trans–cis photoisomerization of the embedded azobenzene moiety was studied, by absorbance and fluorescence spectroscopy techniques. The developed polymeric micellar systems behave as efficient photoresponsive smart nanocarriers.
 Please wait while we load your content...
Please wait while we load your content...

