
Electrical and magnetic properties of the pulsed laser deposited Ca doped LaMnO3 thin films on Si (100) and their electronic structures
Khalid Sultan
*a,
M. Ikram*a,
Sanjeev Gautam bc,
Han-Koo Leed,
Keun Hwa Chaeb and
K. Asokane
bc,
Han-Koo Leed,
Keun Hwa Chaeb and
K. Asokane
aSolid State Physics Lab, Department of Physics, National Institute of Technology, Hazratbal, Srinagar, J & K-190006, India. E-mail: ksbhat.phy@gmail.com; ikram@nitsri.net; Tel: +91-8717000375
bAdvanced Analysis Center, Korea Institute of Science and Technology (KIST), Seoul 136-791, South Korea
cDr S. S. Bhatnagar University Institute of Chemical Engineering and Technology (SSB UICET), Panjab University, Chandigarh, 160-014, India
dBeamline Research Division, Pohang Accelerator Lab (POSTECH), Pohang 790-834, South Korea
eMaterials Science Division, Inter University Accelerator Centre, New Delhi-110067, India
First published on 28th July 2015
Abstract
We report the effect of Ca doping on the structural, electrical, magnetic and electronic properties of stoichiometric La1−xCaxMnO3 (x = 0, 0.3, 0.5 and 0.7) (LCMO) thin films grown on Si (100) using a pulsed laser deposition technique. All these films exhibit a single-phase orthorhombic structure with the space group Pnma. Physical properties, such as surface roughness, grain size, Curie temperature ‘Tc’, activation energy ‘Ea’ and magneto-resistance, were studied as a function of Ca doping. These properties were correlated with the variation of the observed electrical transport and magnetic properties and their electronic structures. The electronic structures of these films were studied by X-ray absorption spectroscopy (XAS) at O K and at Mn L3,2-edges, which indicate an admixture of Mn2+, Mn3+, and Mn4+ ions and also an increase in the density of states with Ca doping. These mixed valence states of Mn ions in LCMO arise due to the doping of Ca in the La sites, which modifies the electrical and magnetic properties.
 Khalid Sultan | Khalid Sultan did his B.Sc at the University of Kashmir with 1st division and obtained his Master's degree in Physics from the University of Kashmir (J & K), India, with 1st division. He has qualified for the prestigious Junior Research Fellowship (JRF) and has qualified the National Eligibility Test (NET) twice in Physical Sciences conducted by CSIR-UGC. He has submitted his Ph.D thesis in Physics (Materials Science) at the National Institute of Technology (NIT) Srinagar, J & K, India. His main research interests include the synthesis and characterization of Mn-based rare earth transition metal oxides and their irradiation study. |
1. Introduction
Even after five decades of research, doping studies of divalent cations such as Ca ions in manganites still fascinate researchers as a result of the evolution of a complex phase diagram that comprises a rich variety of crystallographic, magnetic, and electronic phases.1–3 These compounds exhibit a strong interplay between the spin, charge, orbital, and lattice degrees of freedom. All these properties are dependent on the doping concentration and show drastic changes in the magnetic and transport properties even by subtle modifications in the chemical composition and external parameters such as temperature and pressure.4 LaMnO3 was found to have an orthorhombic structure with the space group Pnma. The deviation of this structure from the cubic structure is usually attributed to the Jahn–Teller instability of the Mn3+ ions. On the other hand, CaMnO3 is thought to have a structure closer to cubic as compared to LaMnO3 (LMO) because the Mn4+ ions do not produce any Jahn–Teller distortion.5 The valency of Mn ions (Mn3+/Mn4+), the oxygen stoichiometry and the ratio between trivalent and divalent cations (R3+/A2+) determine the magnetic and electrical properties in the compound.5 The observed behavior in these compounds is explained on the basis of a double exchange mechanism,6–8 Jahn–Teller distortion,9 lattice, charge and orbital degrees of freedom as well as phase separation.10–12 The strong correlation between the observed transport and the magnetic properties results in novel phenomena with possible future applications.Magneto-electronic devices are often fabricated in the form of thin films. These applications require films of high quality with homogeneous and smooth surface morphology. Many novel techniques have been employed to synthesize LMO thin films such as sol–gel, atomic layer deposition (ALD), molecular beam epitaxy (MBE), and spray pyrolysis.13–17 Several studies have focused on pulsed laser deposition (PLD) for the deposition of thin films of Ca-doped LMO.18 PLD is a well-established technique for depositing high-quality thin films, even those with a complex stoichiometry. Due to the high energy of the laser and the short pulse duration (∼25 ns) the laser-target material interaction is predominantly photonic with only a minimum of thermal heating. Moreover, when the material is fabricated as a thin film, the crystal structure and chemical composition at the surface can have a significant influence.
Commonly, SrTiO3 (STO) and LaAlO3 (LAO) single crystals, having the lattice constants aSTO = 3.91 and aLAO = 3.79 Å, respectively, are used as substrates because their lattices match with that of bulk LMO. Theoretically, this allows the growth of single crystalline and epitaxial films. On the other hand, thin films on Si (aSi = 5.43 Å) are expected to have a large lattice mismatch and thus a polycrystalline nature. Therefore, depositing LMO films on Si causes strain due to large lattice mismatch and surface roughness. It is also well known that the surface composition of rare-earth manganites thin films differs from the bulk sample due to segregation effects.19,20 In the case of LMO, the formal valencies are La3+Mn3+O32−, and for CaMnO3, they are Ca2+Mn4+O32−; thus, (La, Ca) MnO3 should contain both Mn3+ and Mn4+ ions. This mixed valency nature modifies the magnetic and electronic properties of the system. For instance, the parent compounds LaMnO3 and CaMnO3 are antiferromagnetic insulators at low temperatures; however, La0.7Ca0.3MnO3 (LCMO) is a ferromagnetic metal. The mixed valence states of Mn ions modify the electrical and magnetic properties of the LCMO system. To understand the local electronic structure, including the valency of Mn and the hybridization of the Mn 3d–O 2p states, X-ray absorption spectroscopy (XAS) is a most suitable experimental technique and has been used extensively in transition metal oxides.
The present study focuses on the structure, morphology, transport, magnetic and magnetoresistance properties of LCMO films fabricated on Si substrates and the modifications in their electronic structures. The emphasis is on understanding these physical properties and electronic structures to correlate them with the phase diagram of Ca-doped LaMnO3 thin films fabricated on Si substrates.
2. Experimental details
Compounds with the compositions of La1−xCaxMnO3 (x = 0.0, 0.3, 0.5 and 0.7) (hereafter referred as LCMO) were prepared using a conventional solid state reaction technique. Mixed powders of La2O3, Mn2O3 and CaCO3 in required stoichiometric ratios were preheated at 1000 °C for 12 hours, calcined again at 1200 °C for 12 hours and sintered. Thin films of LCMO on Si (100) were prepared by the PLD method. The chamber was cleaned prior to PLD deposition and the base pressure was maintained at ∼1 × 10−5 Torr. A Lambda-Physik LPX210 krypton–fluorine excimer laser (wavelength = 248 nm, pulse length = 20 ns, repetition rate = 10 Hz) was used to deliver a pulsed-laser beam of energy density = 1.8 J cm−2 at the target surface. The target–substrate distance was kept at 5 cm. The substrate was placed at 750 °C during the deposition to ensure highly oriented growth. The ablation was carried out in oxygen at a partial pressure of 330 mTorr. The deposition was carried out for 17 minutes using a laser energy of 200 mJ. After deposition, the films were cooled to room temperature under an ambient oxygen pressure at 20 °C min−1. The film thickness was estimated to be around 70 ± 5 nm using an XP1 Tely step profilometer.The films were characterized by X-ray diffraction (XRD) (Bruker AXS D8 Discover) (Cu-Kα radiation) at room temperature in the 2θ range of 20–80°. The surface morphology was studied using an atomic force microscope (AFM) (Nanoscope E-digital instrument. Inc., USA) in the contact mode. The magnetic measurements were carried out using a 7 Tesla MPMS SQUID-VSM. Temperature dependent magnetization was carried out from 5 K to 300 K under a field of 100 Oe under both field cooled (FC) and zero-field cooled (ZFC) conditions. Electrical resistance R(H) under a magnetic field H was measured from 5 K to 300 K using a standard four-probe technique. All these electrical and magnetic measurements were carried out at IUC, CSR Indore.
X-ray absorption spectra at O K and Mn L3,2 edges were performed at the soft X-ray beamline 10D XAS KIST (Korea Institute of Science and Technology) of the Pohang light source (PLS), S. Korea, by operating at 2.5 GeV with a constant (top-up mode) storage current of 400 mA. The spectra were obtained using the sample drain current mode (TEY) at room temperature, and the vacuum in the experimental chamber was in the low range of 10−9 Torr. The resolution of the spectra was better than 0.6 eV at O K-edge. The data is normalized to the edge-jump at the post-edge region after removing the background at the pre-edge straight region.
3. Results and discussions
3.1 Structural and morphological studies
Fig. 1 shows the glancing incidence XRD (GIXRD) pattern of the LCMO thin films. All these films were polycrystalline without detectable secondary phases and were indexed with the orthorhombic structure of the space group Pnma. Abrashev et al.21 reported that, unlike other perovskite manganites, the average crystal structure of La1−xCaxMnO3 remains orthorhombic in the entire range of Ca substitution at temperatures below ∼700 K. With neutron and X-ray diffraction, Rodriguez et al.22 confirmed that LMO has an orthorhombic structure below ∼800 K. The compositions with x = 0.5 and x = 0.83 can be indexed with the pseudocubic perovskite structure at room temperature and a cubic to tetragonal structural phase transition arises on lowering the temperature, as reported by Zheng et al.23 It has been reported by Banerjee and Krishnan24 that LMO crystallizes into a rhombohedral structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c) with a small distortion. Nagabbushane et al.25 reported that nanocrystalline LMO synthesized by a combustion process is in the cubic phase.
c) with a small distortion. Nagabbushane et al.25 reported that nanocrystalline LMO synthesized by a combustion process is in the cubic phase.
 | ||
| Fig. 1 The GIXRD pattern of LCMO thin films. Inset shows the XRD pattern with the substrate. | ||
On calcination at 900 °C, the structure of Ca-doped manganites changes to the rhombohedral phase, while pristine LMO retains the cubic phase. All these studies show that the crystal structure of LMO is highly sensitive to preparation conditions and that the physical properties are highly dependent on the structure.
Furthermore, the strain (ε) was calculated using the Williamson Hall equation: 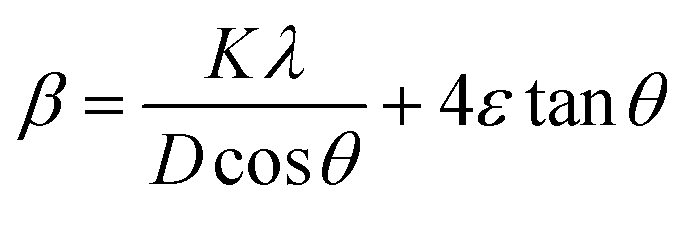 . Rearranging this equation, one obtains
. Rearranging this equation, one obtains

By linear extrapolation of the plot of β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ vs. 4sinθ along the y and x axes, the crystallite size is obtained at the intercept of Kλ/D and the strain (ε) from the slope. Here, β and θ are taken in radians.
cosθ vs. 4sinθ along the y and x axes, the crystallite size is obtained at the intercept of Kλ/D and the strain (ε) from the slope. Here, β and θ are taken in radians.
The lattice parameters, full width at half maximum (FWHM) calculated from peak (121) and strain are presented in Table 1. With Ca doping, it is observed that the calculated strain and FWHM increases but the intensity decreases marginally, indicating changes in the crystallinity of the LCMO thin films. In addition, there was deviation in the lattice parameters.
| Film | a (Å) | b (Å) | c (Å) | FWHM (degrees) | Strain (× 10−1) |
|---|---|---|---|---|---|
| x = 0 | 5.521 | 7.712 | 5.541 | 0.416 | 1.07 |
| x = 0.3 | 5.413 | 7.733 | 5.489 | 0.431 | 1.11 |
| x = 0.5 | 5.425 | 7.669 | 5.431 | 0.425 | 1.09 |
| x = 0.7 | 5.312 | 7.570 | 5.391 | 0.496 | 1.20 |
The cations, La and Ca, have almost identical ionic radii and are randomly substituted on the A site. The size of the ions determines the extent of distortion in the compound. If the size of the A site cation, rA, <a/√2 − ro (here ‘a’ is the cubic lattice parameter and ro is the radius of oxygen ion), the distortion will most probably be due to rigid tilts of oxygen octahedra. On the contrary, if rB < a/2 − ro (rB is the radius of the B site cation), the distortion is most likely to be due to the movement of the B site cation within the oxygen octahedra. The present study follows the former (rA < a/√2 − ro) relation between its contributing ions, thus the distortion probably occurs as a result of the rotation of oxygen octahedra at room temperature. This variation has been attributed to the decrease in the effective ionic radii of La and Mn on Ca doping. The distortion of MnO6 octahedra depends not only on the ionic radii of the dopants but also on the amount of doping.
Fig. 2 shows the AFM images of LCMO thin films. The surface roughness of these films are approximately 1.86 nm, 2.64 nm, 2.27 nm and 2.62 nm for x = 0, 0.3, 0.5 and 0.7, respectively. The values listed in Table 2 suggest that the films are extremely smooth in general. There are very small grains with an average diameter of about 44 nm, 46 nm, 45 nm and 51 nm for x = 0.0, 0.3, 0.5 and 0.7, respectively. The observed grain diameters of these films, which were prepared by PLD, were less compared to those prepared by other groups17 using ALD. This implies that deposition techniques have a considerable role in grain formation. This study shows that with Ca doping, there is a change in the surface roughness and grain sizes of the LCMO films.
 | ||
| Fig. 2 AFM images (size: 1 μm × 1 μm) of LCMO films on Si (100): (a) x = 0.0, (b) x = 0.3, (c) x = 0.5, and (d) x = 0.7. | ||
3.2 Magnetic measurements
The ZFC and FC magnetization as a function of temperature at 100 Oe field are shown in Fig. 3. The large difference between FC and ZFC curves at low temperatures suggests an inhomogeneous mixture of a ferromagnetic and anti-ferromagnetic ordering rather than a distinct long range order. This also indicates the presence of magnetic ordering in the film. The appearance of peaks in the ZFC curves is characteristic of a blocking mechanism, because of the competition between the thermal energy and the magnetic anisotropy energy. The values of magnetic transition temperature ‘Tc’ (the temperature at which the maximum slope dM/dT occurs) for x = 0.0, 0.3 and 0.5 are 140 K, 230 K and 235 K, respectively. The values of the ferromagnetic Curie temperature (Tc), blocking temperature (TB), saturation magnetization (Ms), retentivity (Mr) and coercivity (Hc) for the LCMO films are presented in Table 2. It is observed that the value of Tc increases with Ca doping in the LCMO system up to x = 0.5. For the composition x = 0.7, no sharp magnetic transition is observed. The calculated magnetic parameters Ms, Mr and Hc show an increase with Ca doping, indicating magnetic ordering in films with Ca doping. No anomaly was observed in MT spectra of LMO, as observed by Khanduri et al.;17 they reported that LMO shows broad anomalies at 44 K and 211 K in magnetization vs. temperature curves. The absence of such anomalies may be attributed to a different preparation technique (PLD) and may possibly be due to a different calcination temperature being used in this study during the preparation of films. The shift in transition temperature “Tc” of different doping concentrations may arise due to the presence of lattice disorder and/or strain in the films. The value of the magnetic moment increases progressively with Ca doping but decreases for higher doping (x = 0.7). Magnetization (M), both FC and ZFC, increases rapidly below Tc. At lower temperatures, M becomes saturated in the FC case due to the fully ordered Mn3+ and Mn4+ pairs. However, in the case of ZFC magnetization, M does not become saturated even at lower temperatures. | ||
| Fig. 3 Temperature dependence of ZFC and FC magnetization plots (in the presence of 100 Oe magnetic field) of LCMO films for (a) x = 0.0, (b) x = 0.3, (c) x = 0.5, and (d) x = 0.7. It is observed that the value of Tc increases with Ca doping in the LCMO system up to x = 0.5. For the composition x = 0.7, there is no sharp magnetic transition. | ||
In LCMO, there are three basic phases: insulating-paramagnetic, ferromagnetic (with metallic-like conductivity) and charge-ordered antiferromagnetic insulating phase. The parent compound, LMO, is an antiferromagnetic insulator and Ca doping at La sites results in the phenomenon of colossal magneto-resistance (CMR) for the doping range 0.2 < x < 0.5. CMR occurs in these compositions as a consequence of a rapid shift of the ferromagnetic transition temperature (Tc) to a higher temperature region in the presence of a magnetic field.
The compound La0.5Ca0.5MnO3 shows the coexistence of two totally dissimilar ground states, i.e. ferromagnetic-metallic and antiferromagnetic-charge ordered, which is also consistent with earlier reports.26,27 For the doping value of x = 0.7, the compound is an antiferromagnetic insulator with ordering of doped charge carriers. There is no sharp magnetic transition and the magnetic signal is also very low. Mathur et al.28 reported that when x = 0.7, the crystal structure of manganite interacts strongly with the corresponding magnetic and electronic structures, and any imposed physical discontinuities in the actual samples are likely to create or destroy the delicate phase balance.
The plots of magnetization vs. field (MH) curves at 300 K (Fig. 4) show paramagnetic behavior at room temperature, whereas at 5 K, (Fig. 5, except for the composition of x = 0.7) they show ferromagnetic behavior, which is consistent with M vs. T data discussed above. The magnetic hysteresis loop at room temperature (300 K) is almost paramagnetic. However, below the transition temperature, the films show strong ferromagnetic behavior. It is seen that the value of the saturation magnetization increases up to x = 0.3 and then decreases with doping. For the composition of x = 0.7, the films show an antiferromagnetic nature with ordering of doped charge carriers. The maximum hysteresis is observed for the composition of x = 0.5. The observed large hysteresis is also reported in the literature for these compounds.28,29
 | ||
| Fig. 4 Magnetization vs. field curves at 300 K for LCMO thin films. | ||
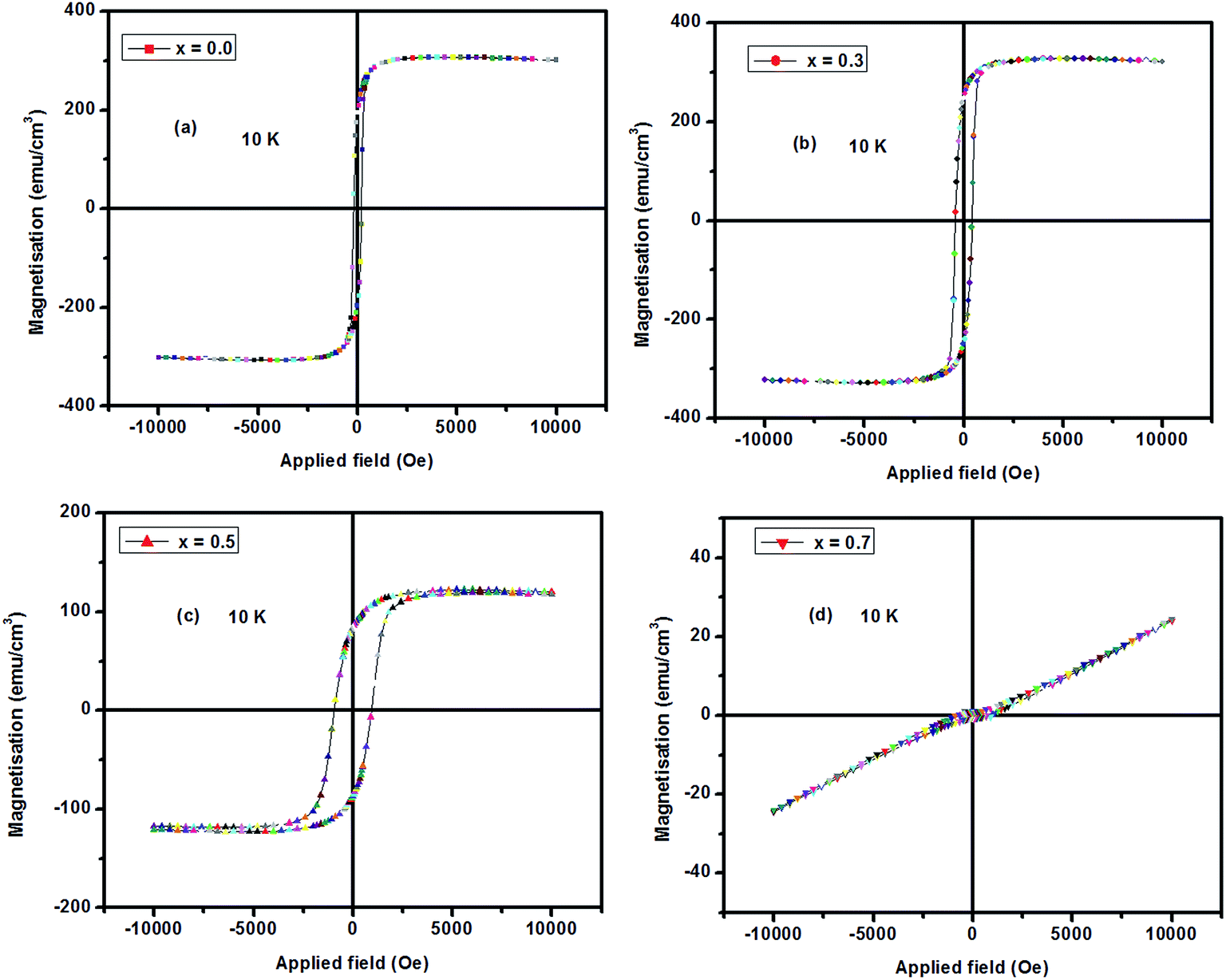 | ||
| Fig. 5 Magnetization vs. field curves at 10 K for LCMO films. The value of saturation magnetization increases up to x = 0.3 and then decreases with doping. | ||
3.3 Electrical transport measurements
The electrical resistivity (ρ) of the thin films, x = 0–0.7, at both H = 0 and H = 8 T as a function of temperature (range, 5–300 K) are shown in Fig. 6. The activation energy (Ea) was calculated from the slope of the curve of logρ vs. 1000/T (K−1) using the relation ρ = ρ0e−Ea/kT and the values are shown in Table 2. Here, ρ0 is the conductivity at infinite temperature, k is Boltzmann's constant and Ea is the activation energy. The pre-factor ρ0 is of the order of 1.5 mΩ, implying that the holes are localized in the paramagnetic phase. This localization is most probably due to the coulombic attraction of the divalent cation or lattice distortion around it. It is clear from Table 2 that the value of Ea first decreases with doping but later increases for higher doping (x = 0.7). The observed trend is consistent with magnetic data, in which magnetization first increases with doping for x = 0.3 and then decreases for higher doping concentrations.
 | ||
| Fig. 6 Temperature dependence of resistivity of LCMO films for (a) x = 0.0, (b) x = 0.3, (c) x = 0.5, and (d) x = 0.7. These films show a decrease in resistivity with Ca doping but the resistivity increases for more highly doped samples (x = 0.7). | ||
All these LCMO thin films are paramagnetic insulators and have an orthorhombic crystal structure at room temperature (300 K).30 There is a decrease in resistivity with Ca doping but the resistivity increases for more highly doped films (x = 0.7). The data from Miller et al.31 shows a sharp increase in the resistivity for both end members: LaMnO3 and CaMnO3. This is reminiscent of the large change in resistivity observed in semiconducting materials when donor or acceptor levels are introduced by doping with small quantities of atoms with a different valency. It is likely that the LMO film used in this investigation contains an excess of oxygen, which introduces a mixed valency into the system (sometimes this process is referred to as ‘self-doping’); this could reduce the resistivity by an order of magnitude. The observed trend in resistivity is consistent with the magnetic data as well as with Ea values. A possible explanation for the high resistivity for x = 0.7 is based on grain boundaries. It is clear from Table 2 that the grain diameter is larger for higher doping concentrations. The observed trend is also consistent with the reports of Edwards et al.,32 where it was found that the grain diameter has considerable dependence on the resistivity of the samples. In the studied polycrystalline samples, well defined grain morphology on a Si substrate was observed. The zero-field resistivity increases systematically with increasing grain size due to enhanced scattering at the grain boundaries.
The field-dependent resistivity (magnetoresistance MR% = (ρ − ρ0/ρ0) × 100) of the films at 5 K, 100 K, 200 K and 300 K are shown in Fig. 7(a)–(d) for films with (a) x = 0.0, (b) x = 0.3, (c) x = 0.5 and (d) x = 0.7.
 | ||
| Fig. 7 Magnetoresistance values for LCMO thin films for (a) x = 0.0, (b) x = 0.3, (c) x = 0.5, and (d) x = 0.7. The value of MR% is maximum (70–80%) for intermediate doping concentrations (x = 0.3 and 0.5) at H = 8 Tesla. | ||
For x = 0.0 and x = 0.7, the MR values are only shown at 200 K and 300 K because the resistivity of these films are too high at lower temperatures. It is observed that at all doping concentrations, resistivity decreases with temperature and is consistent with resistivity data discussed above. The MR also increases with magnetic field for all doping concentrations. The value of MR% is maximum (70–80%) for intermediate doping concentrations (x = 0.3 and 0.5) at H = 8 Tesla. There is a visible similarity in the nature of the change of resistivity with Ca doping to that of the dependence on magnetic Tc. Resistivity decreases sharply with Mn content, reaches a minimum at x = 0.5 and then increases; magnetization also increases with doping, reaches a maximum at x = 0.5 and then decreases. The variation of these transport properties with Ca doping reflects changes in conduction band structure, which are evident from X-ray absorption spectroscopy studies.
3.4 Electronic structure by XAS
To understand the element-specific electronic structure and chemical environment of Mn and O ions in the compound LCMO thin films, XAS studies were carried out. The normalized XAS spectra at O K edge at 300 K is shown in Fig. 8(a). The electronic structure close to the Fermi level (EF) is dominated by Mn 3d and O 2p states in manganites. The O K edge probes the unoccupied density of states with the O 2p symmetry due to dipole selection rules that arise mainly from the hybridization of O 2p states with various states of neighboring atoms, namely, 3d states of Mn as well as the 4d states of La.33 These spectra show main features ‘A’, ‘B’, ‘C’ and ‘D’ at 527.5, 530.6, 534.1 and 541.4, respectively. The peaks ‘A’ and ‘B’ arise from the hybridized O 2p–Mn 3d states and are split into t2g and eg orbitals under the influence of an octahedral crystal field.34 The peak ‘C’ in Fig. 8(a) is attributed to the hybridization of the O 2p orbital with La 5d-states. The broad peak ‘D’ is attributed to the bands of higher energy metallic states, e.g. Mn 4sp and La 6sp bands. The well-resolved peaks marked as ‘A’ and ‘B’ in pure LaMnO3 correspond to a crystal field splitting energy of 10 Dq = ∼2.9 eV for eg and t2g orbitals of the 3d energy level of Mn. All these features are commonly observed in perovskites,35 including nickelates,36 titanates37 and manganites.38,39 | ||
| Fig. 8 (a) Normalized O K-edge XAS spectra of La1−xCaxMnO3 (x = 0.0, 0.3, 0.5, and 0.7) thin films. (b) Difference spectra for the same films at O K edge. It is observed that the intensity of peak ‘B’ increases with Ca concentration, implying a change in density of states. | ||
The changes in the spectral features of the compound LMO with the inclusion of Ca is depicted in the O K-edge difference spectra, as shown in Fig. 8(b). The intensity of peak B provides a measure of the Mn 3d-state occupancy. From Fig. 8(b), it is observed that the intensity of peak ‘B’ increases with Ca concentration. An increase in peak intensity indicates lower Mn 3d-state occupancy, i.e. higher Mn valency. The origin of Mn2+ is interpreted by suggesting a charge disproportionation model based on the instability of Mn3+ with respect to Mn2+ formation via 2Mn3+ → Mn2+ + Mn4+ by Hundley et al.33 and de Jong et al.34 It is also observed from the difference spectra that the density of states increases with Ca doping (the spectral peaks from ‘A’ to ‘D’).
Fig. 9(a) shows XAS at Mn L3,2-edges of LCMO films. Due to the spin–orbit splitting of the 2p core level, 2p → d-symmetry transitions give rise to two edges, namely, the 2p3/2 (L3-edge) at 639 eV and the 2p1/2 (L2-edge) at 650 eV, separated by ∼11 eV. Each spectral edge has sharp peaks due to the large number of conduction band 3d-states. Compared to 2p1/2, a higher number of electrons is available in 2p3/2; therefore, the intensity of the L3-peak is expected to be higher compared to the L2-peak. It has been reported earlier that the energy position of spectral features in the Mn L3,2 XAS study are very sensitive to the oxidation state of Mn atoms and can be used to determine the Mn valency.40–44 The double peak feature of the L3-edge changes as the Ca-concentration increases. The changes in the spectral features of the compound LMO with the inclusion of Ca are depicted in the L3,2-edge difference spectra, as shown in Fig. 9(b). The multiplet feature L3-edge are marked as ‘A1’ and ‘B1’. With an increase in Ca-concentration, there is a change in the mixed valent states of Mn2+/Mn3+ in the system. It is also seen that the L3-edge shifts towards higher energy and there is corresponding modification in the spectral shape, which is suggestive of the fact that the valency of Mn changes with Ca doping.42 It is also observed from Fig. 9 that the peak at 638.8 eV, shown by ‘A1’, is typical for Mn2+ and the peak around 640.3 eV is typical for Mn3+. Therefore, if Mn is in a mixed valency state, a mixture of Mn2+ and Mn3+, a double peak Mn L3-feature is expected. With these insights, one concludes that LCMO films contain both Mn2+ and Mn3+ ions. Due to this mixed valence state of Mn induced by the introduction of Ca in the LCMO system, a multichannel double exchange mechanism (Mn2+–O–Mn3+ and Mn3+–O–Mn4+) may be favored. This implies that the mixed valence state of Mn ions is responsible for modifying the magnetic and other transport properties of the LCMO system.
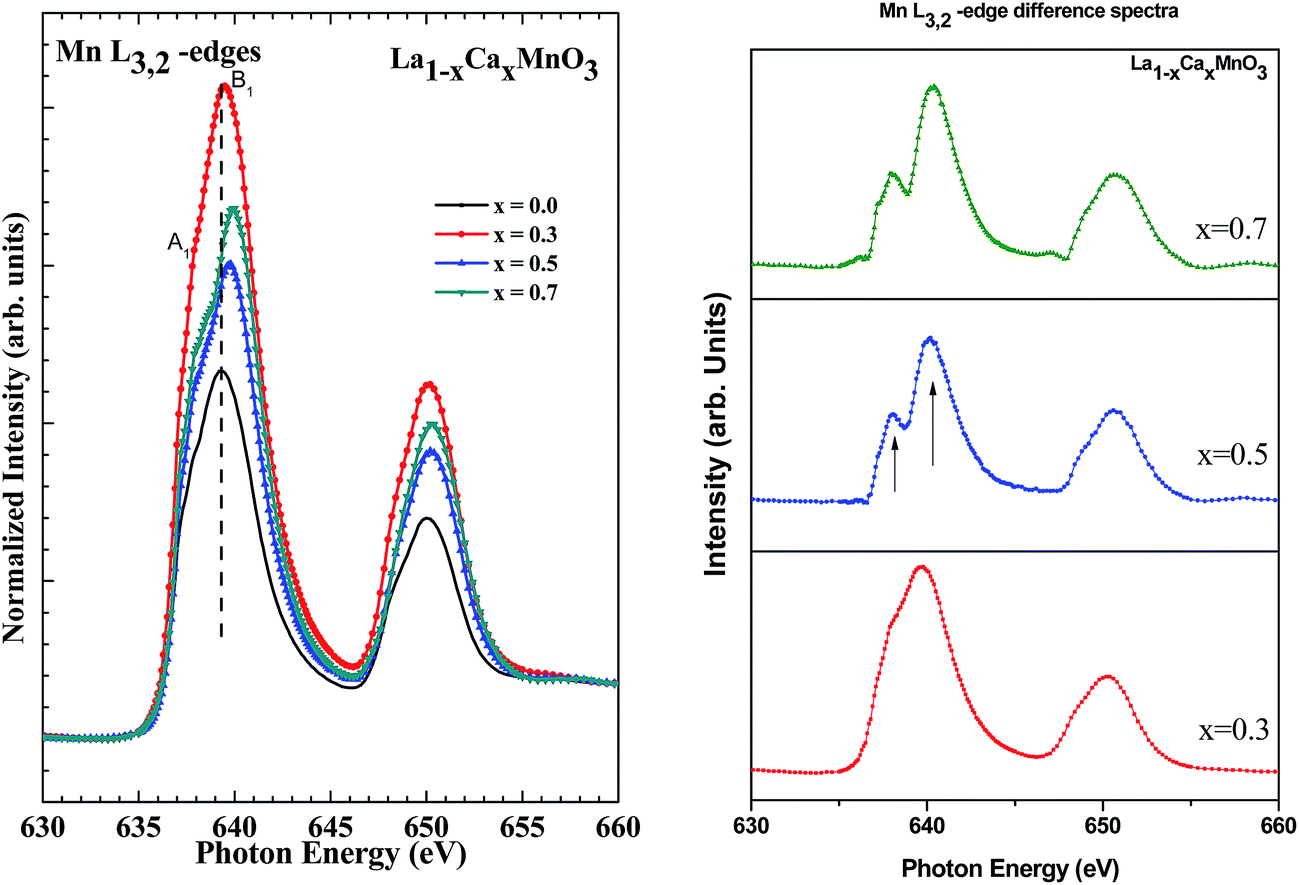 | ||
| Fig. 9 (a) Normalized Mn L3,2-edge XAS spectra of LCMO thin films. (b) Difference spectra for Mn L3,2-edge. Note that the Mn L3-edge shifts towards higher energy and there is corresponding modification in the spectral shape, which is suggestive of the fact that the valency of Mn changes with Ca doping. | ||
The overall magnetic and electrical behavior depends on the interactions between the different Mn ions. Four counteracting processes determine the behavior of the Mn 3d valence electrons. These are double exchange interaction, super exchange interaction, Jahn–Teller effect and charge ordering. Parameters, such as temperature, doping, strain and crystal structure, determine the dominant mechanism. Deformation of the oxygen octahedron causes energy differences within the otherwise degenerate t2g and eg electron states. The orbitals that are compressed by the deformation gain energy, while the orbitals that are elongated by the deformation loose energy. When the eg shell is half filled, it is energetically favorable to create a deformation because one of the orbitals will lose energy. The electrons in the eg shell will then occupy this lower energy state. Creating a deformation of the oxygen octahedrons without altering the overall crystal lattice leads to typical Jahn–Teller distortions. During the double exchange interaction, two neighboring Mn ions and their connecting O ion play an important role. The Mn ion in this system has one unoccupied 3d orbital. Thus, one electron from the O 2p orbital tunnels to this Mn 3d orbital. With the formation of the unoccupied O 2p orbital, the electron from the other Mn 3d orbital moves. The net result is the movement of one electron from one Mn ion to another Mn ion. The qualitative image of this behavior in the mixed-valent manganite, namely, La0.5Ca0.5MnO3, has been attributed to the concept of Zener double exchange.6,30
The influence of substrate strain is the main factor that distinguishes manganite thin films from bulk ceramic samples. LaAlO3 is a commonly used substrate that has least lattice mismatch to LCMO at room temperature and hence low strain. The effect of various substrates, as well as film thickness, on MR has been studied by Jin et al.,45 and it was observed that the strain effect, expressed through the MR, is most pronounced for epitaxial films. It is speculated that stress is most effectively propagated through the film thickness in the absence of grain boundaries. For the LaAlO3 substrate, MR is maximized for a film thickness of 1000 Å. In this study, the fact that LCMO films were deposited on Si (100), which has a larger lattice mismatch with LCMO at room temperature (as discussed in the Introduction), may induce additional strain in the films. Pristine LMO is in an anti-ferromagnetic insulating (AFI) state up to 150 K and then changes into a paramagnetic state (PM). The film with the composition of x = 0.3 was found to be in the ferromagnetic region up to 240 K, above which it changed into a PM state. The film with composition of x = 0.5 was found to be in a FM state up to 245 K, above which they changed to a PM state. The films with a composition of x > 0.5 (x = 0.7) were found to be in a mixture of charge ordered insulating (COI) and AFI states. Ramirez46 reported the Tc for concentration of x = ∼0.3 to be around 220 K, while in this study, it was observed to be around 240 K. Therefore, the large lattice mismatch between Si and LCMO results in a change in the phase diagram of the LCMO system when it is deposited on Si substrates. The mixed valent LCMO exhibits a large variety of states. Such variation occurs because of the presence of various competing and cooperating interactions that involve the charge, orbital, spin and lattice degrees of freedom. In the present study, the interactions are described using a simple ionic image of Mn 3d orbitals. The Mn4+ ion contains three electrons in the t2g state, which are localized and have collinear spins because of the large Hund's coupling. With Ca doping in the parent compound LaMnO3, the extra electron goes into the eg orbitals. There is also a large Hund's coupling between the eg electron and the immobile t2g spin. As a result, the metallic ground state in manganites is also ferromagnetic. In the LCMO system, oxygen 2p orbitals play an essential role and are strongly hybridized with the Mn orbitals. The image described in the LCMO system can be understood as a parameterization in which the oxygen degrees of freedom are integrated and new effective degrees of freedom are introduced by Mn 3d orbitals.
4. Conclusion
Thin films of LCMO were deposited on a Si (100) substrate by the PLD technique and were characterized by various techniques to understand their structural, morphological, electrical, magnetic properties and electronic structures. From the magnetic measurements, it was observed that the value of Tc increases with Ca doping in the LCMO system up to x = 0.5 and there was no sharp magnetic transition at the composition of x = 0.7. The value of the magnetic moment progressively increases with Ca doping but decreases for higher doping (x = 0.7). The value of the saturation magnetization increases up to x = 0.3 and then decreases with doping. The film with the composition of x = 0.7 shows an antiferromagnetic nature with ordering of doped charge carriers. The maximum hysteresis is observed with the composition of x = 0.5. The MR is found to increase with field for all the doping concentrations. The value of MR% is maximum (70–80%) for intermediate doping concentrations (x = 0.3 and 0.5) at H = 8 Tesla. Spectroscopic studies using XAS indicate an increase in Mn valency, confirming an admixture of Mn2+, Mn3+, and Mn4+; these also suggest that the density of states increases with Ca doping. The phase diagram of the Ca-doped LMO exhibits significant changes with doping concentration. The pristine LMO exhibits an AFI state up to 150 K and then changes into the PM. As the density of states increases due to doping at x = 0.3, LCMO films show ferromagnetic characteristics up to 240 K, then change into the PM state. At x = 0.5, these films are found to be in the FM state up to 245 K, and then change to the PM state. The films with x > 0.5 (x = 0.7) were found to be in a mixture of COI and AFI states. Depending upon the doping concentration, different phases evolve, which are strongly related to their electronic structure induced by the Ca dopant.Acknowledgements
Authors (K. S. and M. I.) thank Dr R. J. Choudhary, Dr R. Rawat, and Dr V. R. Reddy, IUC, CSR Indore, for magnetic, transport and XRD measurements, respectively, and the Director of IUAC, New Delhi, for necessary experimental facilities. They also thank the Director, NIT Srinagar for encouragement and support.References
- R. Korotana, G. Mallia, Z. Gercsi, L. Liborio and N. M. Harrison, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 205110 CrossRef.
- J. A. Turcaud, A. M. Pereira and L. F. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 134410 CrossRef.
- C. Raisch, C. Langheinrich, R. Werner, R. Kleiner, D. Koelle, M. Glaser, T. Chassé and A. Chassé, J. Appl. Phys., 2013, 113, 063511 CrossRef PubMed.
- L. Yu-Kuai, Y. Yue-Wei and L. Xiao-Guang, Chin. Phys. B, 2013, 22, 087502 CrossRef PubMed.
- A. J. Mills, Nature, 1998, 392, 147 CrossRef.
- C. Zener, Phys. Rev., 1951, 81, 440 CrossRef CAS.
- P. W. Anderson and H. Hasegawa, Phys. Rev., 1955, 100, 675 CrossRef CAS.
- P. H. de Gennes, Phys. Rev., 1960, 118, 141 CrossRef CAS.
- J. B. Goodenough, Annu. Rev. Mater. Sci., 1998, 28, 1 CrossRef CAS.
- M. Uehara, S. Mori, C. H. Chen and S. W. Cheong, Nature, 1999, 399, 560 CrossRef CAS.
- Y. Tokura and N. Nagaosa, Science, 2000, 288, 462 CrossRef CAS.
- A. Moreo, S. Yunoki and E. Dagotto, Science, 1999, 283, 2034 CrossRef CAS.
- A. Kleine, Y. Luo and K. Samwer, Europhys. Lett., 2006, 76, 135 CrossRef CAS.
- C. Aruta, et al., J. Appl. Phys., 2006, 100, 023910 CrossRef PubMed.
- G. Kartopu and M. E. Souni, J. Appl. Phys., 2006, 99, 033501 CrossRef PubMed.
- P. Orgaini, C. Aruta, R. Ciancio, A. Galdi and L. Maritato, Appl. Phys. Lett., 2009, 95, 013510 CrossRef PubMed.
- H. Khanduri, et al., J. Phys. D: Appl. Phys., 2013, 46, 175003 CrossRef.
- H. Y. Hwang, S. W. Cheong, P. G. Radaeli, M. Marezio and B. Batlogg, Phys. Rev. Lett., 1995, 75, 914–917 CrossRef CAS.
- H. Dulli, P. A. Dowben, S.-H. Liou and E. W. Plummer, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R14629 CrossRef CAS.
- J. Choi, J. Zhang, S.-H. Liou, P. A. Dowben and E. W. Plummer, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 13453 CrossRef CAS.
- M. V. Abrashev, J. Backstram, L. Borjesson, V. N. Popov, R. A. Chakalov, N. Kolev, R. L. Meng and M. N. Iliev, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 184301 CrossRef.
- J. Rodriguez-Carvajal, M. Hennion, F. Moussa, A. H. Moudden, L. Pinsard and A. Revcolevschi, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, R3189 CrossRef CAS.
- R. K. Zheng, C. F. Zhu, I. Q. Xie and X. G. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 63, 020427 CrossRef.
- R. V. Krishnan and A. Banerjee, Solid State Phys., 1998, 41, 431 Search PubMed.
- R. M. Nagabbushane, G. T. Chandrappa, R. P. Sreekanth Chakradhar and K. P. Ramesh, Solid State Phys., 2004, 49, 293 Search PubMed.
- A. Moreo, S. Yunoki and E. Dagotto, Science, 1999, 283, 2034–2040 CrossRef CAS.
- N. D. Mathur and P. B. Littlewood, Solid State Commun., 2001, 119, 271–280 CrossRef CAS.
- M. Uehara, S. Mori, C. H. Chen and S. W. Cheong, Nature, 1999, 399, 560–563 CrossRef CAS.
- P. Levy, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 6437–6441 CrossRef CAS.
- S. W. Cheong and H. Y. Hwang, in Colossal Magnetoresistive Oxides – Monographs in Condensed Matter Science, ed. Y. Tokura, Gordon & Breach, Reading, 2000 Search PubMed.
- R. C. Miller, R. R. Heikes and R. Mazelsky, J. Appl. Phys., 1961, 32, 3302 Search PubMed.
- P. P. Edwards and M. J. Sienko, Phys. Rev. B: Solid State, 1978, 17, 2575 CrossRef CAS.
- M. F. Hundley and J. J. Neumeier, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 11511 CrossRef CAS.
- M. P. de Jong, I. Bergenti, V. A. Dediu, M. Fahlman, M. Marsi and C. Taliani, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 014434 CrossRef.
- P. A. van Aken, B. Liebscher and V. J. Styrsa, Phys. Chem. Miner., 1998, 25, 494 CrossRef CAS.
- D. D. Sarma, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14238 CrossRef CAS.
- K. Asokan, et al., J. Phys.: Condens. Matter, 2001, 13, 11087 CrossRef CAS.
- M. Abbate, D. Cruz, G. Zampieri, J. Briatico, M. T. Causa, M. Tovar, A. Caneiro, B. Alascio and E. Morikawa, Solid State Commun., 1997, 103, 9 CrossRef CAS.
- M. Abbate, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 4511 CrossRef CAS.
- L. A. J. Garvie and A. J. Craven, Phys. Chem. Miner., 1994, 21, 191 CrossRef CAS.
- J. Kawai, Y. Mizutani, T. Sugimura, M. Sai, T. Higuchi, Y. Harada, Y. Ishiwata, A. Fukushima, M. Fujisawa, M. Watanabec, K. Maeda, S. Shin and Y. Gohshi, Spectrochim. Acta, Part B, 2000, 55, 1385 CrossRef.
- S. P. Cramer, F. M. F. de Groot, Y. Ma, C. T. Chen, F. Sette, C. A. Kipke, D. M. Eichhorn, M. K. Chan, W. H. Armstrong, E. Libby, G. Christou, S. Brooker, V. McKee, O. C. Mullins and J. C. Fuggle, J. Am. Chem. Soc., 1991, 113, 7937 CrossRef CAS.
- H. K. Schmid and W. Mader, Micron, 2006, 37, 426 CrossRef CAS PubMed.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5459 CrossRef CAS.
- S. Jin, T. H. Tiefel, M. McCormack, H. M. O'Bryan, L. H. Chen, R. Ramesh and D. Schurig, Appl. Phys. Lett., 1995, 67, 557 CrossRef CAS PubMed.
- A. P. Ramirez, J. Phys.: Condens. Matter, 1997, 9, 8171 CrossRef CAS and references therein.
| This journal is © The Royal Society of Chemistry 2015 |