
Anti-stacking dense conversion of solid organic sodium salt particles into graphene with excellent electrode performance†
H. J. Cuiab,
Y. Y. Zhuab,
J. F. Zheng*a,
S. P. Jiaa,
Z. J. Wanga and
Z. P. Zhu*a
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, China
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China. E-mail: zhengjf@sxicc.ac.cn; zpzhu@sxicc.ac.cn
First published on 22nd June 2015
Abstract
Graphene frameworks can be directly formed by a rapid decomposition of common solid organic sodium salts, during which the gases simultaneously produced play a crucial role in resisting the inter-graphene stacking behaviour through in situ filling between the growing graphene films. The framework structure endows the graphene with excellent electrode performance.
The versatile and supreme properties of graphene have highlighted its great potential for revolutionizing numerous existing materials.1–7 The application of graphene is essentially driven by the progress in its mass production.1 Actually, graphene, as single or few-layer graphite crystals, is rather common structurally, with a hexagonal network structure built by thermodynamically the most stable sp2-hybridized carbon atoms. Although nearly all carbon-containing compounds can be converted into sp2-carbon species, the first synthesis of this common structure was achieved just ten years ago by the physical exfoliation of graphite crystals8 and their mass chemical synthesis still meets a great challenge. This situation is certainly derived from the inherent π–π interaction of graphene films, which normally drives graphene to stack as thick graphite crystals. The current top-down strategy for graphene of exfoliating it from graphite crystal is just based on a compelling breakage of the π–π interactions by physical forces,8,9 molecule intercalation,10 or by oxidative intercalation.11 Although the oxidative exfoliation was fully proven as efficient and scalable,1 the required lengthy oxidation–reduction procedure and the involved environmental problems would raise production costs greatly. Bottom-up chemical synthesis of graphene from atomic or molecular carbon species has been intensively studied and has had much successes, such as the SiC decomposition based epitaxial growth,12,13 the chemical vapor deposition (CVD) of hydrocarbons,14–17 the carbonization of organic polymer thin films,18 and the polymerization and cyclodehydrogenation of molecular monomers.19,20 These chemical approaches have unique advantages in controlling the size, thickness, and structure of graphene films for diverse applications,21–25 but they are difficult to support for mass production because the baffling π–π stacking problem was not resolved properly, just bypassed via a strict control of product quantity (performing at quite low carbon density). Additionally, in order to direct two-dimensional (2D) growth of graphene, special substrates were usually employed,14–18 which additionally raises synthesis costs because the subsequent graphene transfer or substrate removal is difficult.26,27 Here we show that the rapid decomposition of solid particles of organic sodium salts is capable of directly converting them into core–shell particles constructed of graphene framework (GF) shells and Na2CO3 cores (Fig. 1a). Pure GFs can be readily obtained via removal of the Na2CO3 cores by simply washing with water. In this solid conversion process, the π–π stacking behavior of the growing graphene is spontaneously restrained, assisted by the gases produced simultaneously. Owing to the well-built three-dimensional (3D) framework structure and the oxygen-containing characteristics, the GFs have unique advantages in facilitating quasi-isotropic electron conduction, promoting ion transport and catalyzing molecule or ion reactions. These mean the GFs display good electrode performance. In quantum dot sensitized solar cells (QDSSCs), the photovoltaic efficiency (η) for the GF electrode is 3.57%, much higher than that (2.58%) for a RGO electrode. In supercapacitors, the specific capacitance of the GF electrode is 210 F g−1 at a current density of 0.5 A g−1, which is much better than that of the RGO electrode (150 F g−1).
 | ||
| Fig. 1 The formation of GFs. (a) Schematic of the conversion of solid organic salt particles into GFs. (b) SEM images of the original NaAc particles. (c) As-prepared popcorn-like GF particles with Na2CO3 cores, obtained from a fast decomposition of NaAc particles at 1200 °C. (d) A single GF particle enlarged. (e) GFs purified by removing the Na2CO3 cores. (f) TEM image of the GFs. The inset is a typical HRTEM image. (g) A representative AFM image of the GFs. | ||
In the first demonstration, 1.5 g of as-purchased solid sodium acetate (NaAc) particles (Fig. 1b) loaded in a small ceramic boat were allowed to rapidly decompose in a tubular quartz reactor that was preheated to 1200 °C in an argon stream (see ESI for materials and methods†). After a short time of reaction, typically one minute, the resulting black solids were moved out of the high-temperature zone and allowed to gradually cool to room temperature. Scanning electron microscopy (SEM) images show that the as-prepared product displays popcorn-like particles (Fig. 1c), which are usually linked together into hundreds of particles. The sizes (2–8 μm) of the particles are several-fold greater than the original NaAc sizes (1–3 μm), reflecting a significant volumetric expansion induced by the reaction. From the enlarged images, we observed that the particle surfaces consist of thin graphene films, which display a fine framework structure (Fig. 1d). Elemental analysis revealed that the particles also contain large amounts of sodium and oxygen atoms (Fig. S1†), which exist as a nitrite structure of Na2CO3 crystals, as indicated by X-ray diffraction analysis (Fig. S2†). Accordingly, the as-prepared particles were washed with water fully to remove the Na2CO3, and pure GFs were readily obtained. They display a hierarchical cellular structure, containing additional micrometer-scale macropores (Fig. 1e), which are certainly left behind by the Na2CO3 removal. The observations from both transmission electron microscopy (TEM) and atomic force microscopy (AFM) reveal that the graphene has a few layers (Fig. 1f and g).
The Raman spectrum of the GFs exhibits a sp2-hybrid-characterized G band, a defect-derived D band, and a broad 2D band (Fig. S3†), similar to the spectra of the thermally or chemically reduced graphene oxides.11,28,29 The X-Ray diffraction (XRD) pattern shows obvious differences between the GFs and graphite (Fig. S4†). X-Ray photoelectron spectroscopy (XPS) revealed that about 8.8 at% of oxygen atoms are involved in the GFs and are mainly structured as C–O single bonds (Fig. S5†), which agrees with the result from infrared analysis (Fig. S6†). The oxygen-containing characteristic is also similar to that obtained from the oxidative exfoliation of graphite and subsequent reduction.30 It is beneficial for the functionalized modification and applications of graphene.31–33 N2 absorption measurements (Fig. S7†) showed that the GFs obtained from NaAc exhibit a BET special surface area of about 224.5 m2 g−1. In addition, the obtained GFs have good thermostability. After post-annealing at 1200 °C for 1 h, the GFs still keep their well-constructed framework structure (Fig. S8†).
The above data indicate that solid NaAc particles can be facilely converted into cellular graphene particles. Recently, there were some excellent reports on the synthesis of 3D graphene.34–40 However, our method is quite different from those methods in many aspects. A detailed comparison is given in Table S1.† Compared with those methods, our method is simple, green, cheap and time-saving, which makes it quite possible to use for large scale synthesis and applications.
What is surprising is that the formed thin graphene films do not stack as bulk graphite crystals in such a circumstance with extremely high carbon density (about 35.4 mmol cm−3 for NaAc). There must be a native power involved in the reaction system to resist the π–π interactions in real time. This power is likely associated with the gases (CO2, CO, H2O, H2, and CH4) formed continuously during the NaAc decomposition. In view of the thermal diffusion limitation within the micrometer-sized NaAc particles under the fast heating conditions, predictably, the gas generation from NaAc decomposition and the graphene growth from the carbon species would occur simultaneously in the short reaction duration. When the gas generation rate (proportional to the rate of NaAc decomposition) is faster than the rate of gas diffusion, a local high-pressure environment would be created inside the intermediate particles, in which the growing graphene films would be segregated in real time by intercalation of the dense gases until they are cross linked and stabilized as 3D frameworks. This gas-mediated anti-stacking effect closely links with the expansion behaviour observed during the particle conversion and is somewhat analogous to the situation involved in the thermal exfoliation of graphite oxides reported previously,28,29 where quick decomposition of the rich oxygen-containing groups creates a transient high-pressure circumstance inside the graphite oxide particles, which drives graphite exfoliation as thin graphene films. More evidence for the gas-mediated anti-stacking effect can be extracted from the following experimental observations.
Fast heating of NaAc at a high temperature, which mostly determines the rate of NaAc decomposition, is required for graphene formation. Although 2D carbon growth can also be achieved using a temperature-controlled (10 °C min−1) gradual heating of NaAc particles, the resulting carbon sheets are heavily stacked, with a thickness up to hundreds of nanometers (Fig. S9†). At relatively low temperatures such as 700 °C, carbon species form only using a prolonged reaction time (about 4 min) under the fast heating conditions and exhibit littered carbon debris as thick as tens of nanometers (Fig. S10†). At temperatures above 800 °C, GFs can readily form (Fig. S11 and S12†) and obviously show a thickness decline with increasing temperature (Fig. 2a). This temperature effect can be well explained by the gas-mediated anti-stacking effect because temperature elevation speeds up NaAc decomposition and gas generation (Fig. 2b, Fig. S13†), which will raise the local gas density and pressure inside the intermediate particles and thus resist the inter-graphene stacking behaviour more efficiently.
 | ||
| Fig. 2 Dependence of the NaAc decomposition temperature on (a) graphene thickness and (b) the release of CO2, which was measured in real time using mass spectrometry at 44 amu fragments. | ||
We also performed fast decomposition of the solid particles of other organic sodium salts, such as propionate (NaPr), butyrate (NaBu), succinate (NaSu), and citrate (NaCi). For NaPr and NaBu, which have a relatively lower content of –COO groups and exhibit a lower rate of gas generation in comparison with NaAc (Fig. S14†), the formation of thin graphene (Fig. S15†) requires higher temperatures such as 1200 °C to drive the decomposition and gas generation fast enough for effective resistance to the π–π interactions. For NaSu and NaCi, which have a relatively high content of –COO groups and intrinsically can release more CO2, they can be readily converted into uniform popcorn-like graphene particles (Fig. 3a and b) even at relatively low temperatures such as 1000 °C, and pure cellular GFs can be obtained after removal of the concomitant Na2CO3 (Fig. 3c and d). These observations suggest a generality of the present direct solid conversion method for graphene synthesis and give additional support for the gas-mediated anti-stacking effect. Furthermore, we noticed that the sodium involved in the starting material plays a crucial catalytic role in the graphene growth, at least, in the local fixing of the carbon species as solid carbon because no solid substrate is left behind from the fast decomposition of the solid particles of succinic acid or citric acid under the same conditions.
 | ||
| Fig. 3 From NaSu and NaCi to GF particles. (a and b) Popcorn-like GF particles as-prepared from fast decomposition of NaSu and NaCi, respectively. (c and d) The GFs purified from the popcorn-like particles shown in (a) and (b), respectively. | ||
The practical application of graphene substances in a condensed state requires an inhibition of their re-stacking behaviour, such as the morphological modulation of them as curved or 3D framework structures.29,34,36,41,42 An additional superiority of 3D GFs over the 2D counterparts can be found when they are used as functional electrodes such as those in solar cells and supercapacitors.43–46 In QDSSCs, good conductivity, catalytic ability and ion transport ability of the counter electrode (CE) are required to speed up the reduction of the electrolyte ions, frequently Sx2− ions.47,48 We have compared the GF electrode with an electrode of 2D graphene (RGO, obtained from chemical reduction of 2D graphene oxides11) in their impedance behaviour in S2−/Sx2− electrolytes (Fig. 4a), measured from symmetrical dummy cells comprising two identical electrodes (Fig. S16†). By fitting the Nyquist plots with an equivalent circuit model, the series resistance (Rs) and charge transfer resistance (Rct) at the electrode–electrolyte interface and the Nernst diffusion impedance (ZN) are extracted, as presented in Table S2.† Obviously, the Rct, and ZN values of the GF electrode are both much lower than those of the RGO electrode, indicating that multiple promotion effects are involved for the GFs. Unlike anisotropic 2D graphene films that tend to arrange parallel to the electrode surface and display poor c-axis electron conduction, the 3D framework structure has quasi-isotropic properties and can provide multi-direction channels for electron conduction. Furthermore, the cellular structure of the GFs can greatly facilitate the diffusion of electrolyte ions and provides more accessible surfaces and active sites for the catalytic reduction of Sx2− ions, which closely correlates with its low Rct value.43,49
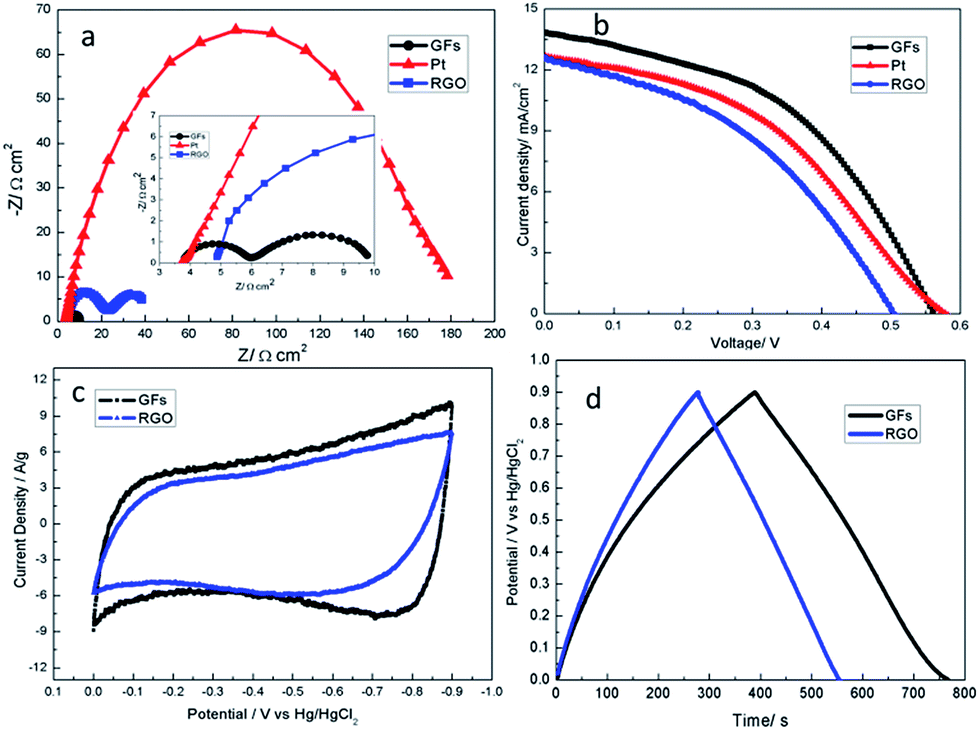 | ||
| Fig. 4 The electrochemical properties of the GF and RGO-modified electrodes. (a) Nyquist plots of the symmetrical cells at a bias of 0.0 V under dark conditions. Inset: a magnified version of the high frequency region. (b) Photocurrent density–voltage curves of the QDSSCs with GF, RGO and Pt counter electrodes, measured at 100 mW cm−2 (AM 1.5). (c) CV curves of the GF and RGO electrodes measured at 50 mV s−1 in 6 M KOH. (d) Galvanostatic charge–discharge curves of the GF and RGO-modified electrodes at a current density of 0.5 A g−1 in 6 M KOH. | ||
The excellent basic properties of the GFs mean a potential to be greater than planar graphene when used as CE materials in QDSSC systems. As tested using packaged cells (see ESI for materials and methods†), the photovoltaic efficiency (η) for the GF electrode is 3.57%, much higher than that (2.58%) for the RGO electrode (Fig. 4b, Table S2†), and similar to the situations reported previously.37 Compared with the platinum (Pt) CE, the GF electrode shows a higher η and exhibits lower Rct and ZN (Table S2†), revealing that the GF electrode has excellent catalytic and ionic transport abilities for S2−/Sx2− electrolytes, which makes it a promising candidate for the counter electrode in QDSSCs.
Due to the framework structure, 3D graphene can also be favorable for application in supercapacitors.46,47 Therefore, the electrochemical energy storage performance of the GF electrode without conductive additives was studied. As shown in Fig. 4c, compared with the cyclic voltammetry (CV) curve of the RGO-modified electrode, the CV curve of the GF-modified electrode had a better rectangular shape, indicating good capacitance behavior. The specific capacitance of the two kinds of electrodes was measured using galvanostatic charge–discharge tests in a three-electrode system. As calculated from the discharge curve (Fig. 4d), the specific capacitance of the GF-modified electrode is 210 F g−1 at a current density of 0.5 A g−1, which is much better than that of the RGO-modified electrode (150 F g−1). The improved capacitance behavior of the GF-modified electrode may be attributed to the framework structure of the GFs, which can provide multi-direction channels for electron conduction and more transport pathways for the diffusion of electrolyte ions. This analysis can be verified by their impedance behavior (Fig. S17 and Table S3†). In addition, the GF-modified electrode also displayed good cycling stability and retained over 92% of its capacitance over 2000 cycles at a current density of 2 A g−1 (Fig. S18†).
Conclusions
In summary, we demonstrated that graphene with a cellular framework structure can be directly and densely formed from solid organic salt particles by a quick thermal decomposition, during which the inherent inter-graphene π–π interactions are effectively restrained in real time by an intercalation effect of the large amount of gases produced locally. Although the graphene synthesized at the present stage is rich in structural defects, its unique cellular framework structure and the oxygen-containing characteristics, as demonstrated, endow it with unique advantages in facilitating quasi-isotropic electron conduction, promoting ion transport, and catalyzing molecular or ionic reactions, which are intrinsically important in QDSSCs and supercapacitors. The η for the GF electrode in QDSSCs is 3.57%, much higher than that (2.58%) for the RGO electrode. The specific capacitance of the GF electrode in supercapacitors is 210 F g−1 at a current density of 0.5 A g−1, which is much better than that of the RGO-modified electrode (150 F g−1). Particularly, after overall consideration of the simple, green, cheap and time-saving synthesis method, the GFs are a promising and competitive electrode material. The knowledge provided on the gas-mediated anti-stacking effect might lead to new strategies for the synthesis of both graphene and other nanosheet materials.Acknowledgements
The authors gratefully acknowledge financial support from the Natural Science Foundation of Shanxi Province (2012011020-1) and Coal Base Key Scientific and Technological Program of Shanxi Province (MC2014-01).Notes and references
- K. S. Novoselov, V. I. Fal’ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192 CrossRef CAS PubMed.
- V. Chabot, D. Higgins, A. Yu, X. Xiao, Z. Chen and J. Zhang, Energy Environ. Sci., 2014, 7, 1564 CAS.
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Adv. Mater., 2014, 26, 849 CrossRef CAS PubMed.
- B. Luo, S. Liu and L. Zhi, Small, 2012, 8, 630 CrossRef CAS PubMed.
- Y. Li, G. Sheng and J. Sheng, J. Mol. Liq., 2014, 199, 474 CrossRef CAS PubMed.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454 CrossRef CAS PubMed.
- R. K. Upadhyay, N. Soin and S. S. Roy, RSC Adv., 2014, 4, 3823 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- I. Y. Jeon, H. J. Choi, S. M. Jung, J. M. Seo, M. J. Kim, L. Dai and J. B. Baek, J. Am. Chem. Soc., 2013, 135, 1386 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun’Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad and P. N. First, J. Phys. Chem. B, 2004, 108, 19912 CrossRef CAS.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS PubMed.
- Y. Lee, S. Bae, H. Jang, S. Jang, S.-E. Zhu, S. H. Sim, Y. I. Song, B. H. Hong and J.-H. Ahn, Nano Lett., 2010, 10, 490 CrossRef CAS PubMed.
- A. Reina, S. Thiele, X. Jia, S. Bhaviripudi, M. S. Dresselhaus, J. A. Schaefer and J. Kong, Nano Res., 2009, 2, 509 CrossRef CAS.
- Z. Sun, Z. Yan, J. Yao, E. Beitler, Y. Zhu and J. M. Tour, Nature, 2010, 468, 549 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Mullen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- Y. Z. Tan, B. Yang, K. Parvez, A. Narita, S. Osella, D. Beljonne, X. Feng and K. Mullen, Nat. Commun., 2013, 4, 2646 Search PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. Ferrari, Nat. Photonics, 2010, 4, 611 CrossRef CAS PubMed.
- J. R. Miller, R. Outlaw and B. Holloway, Science, 2010, 329, 1637 CrossRef CAS PubMed.
- R. Kou, Y. Shao, D. Mei, Z. Nie, D. Wang, C. Wang, V. V. Viswanathan, S. Park, I. A. Aksay and Y. Lin, J. Am. Chem. Soc., 2011, 133, 2541 CrossRef CAS PubMed.
- J. Kang, D. Shin, S. Bae and B. H. Hong, Nanoscale, 2012, 4, 5527 RSC.
- L. Gao, G.-X. Ni, Y. Liu, B. Liu, A. H. Castro Neto and K. P. Loh, Nature, 2013, 505, 190 CrossRef PubMed.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud’homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535 CrossRef CAS PubMed.
- J. Yan, J. Liu, Z. Fan, T. Wei and L. Zhang, Carbon, 2012, 50, 2179 CrossRef CAS PubMed.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845 CrossRef CAS PubMed.
- T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2010, 5, 436 CrossRef PubMed.
- Y. Xu, Z. Liu, X. Zhang, Y. Wang, J. Tian, Y. Huang, Y. Ma, X. Zhang and Y. Chen, Adv. Mater., 2009, 21, 1275 CrossRef CAS PubMed.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490 CrossRef CAS PubMed.
- Z. P. Chen, W. C. Ren, L. B. Gao, B. L. Liu, S. F. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS PubMed.
- Z. S. Wu, Y. Sun, Y. Z. Tan, S. Yang, X. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 19532 CrossRef CAS PubMed.
- Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371 CrossRef CAS PubMed.
- C. M. Chen, Q. Zhang, C. H. Huang, X. C. Zhao, B. S. Zhang, Q. Q. Kong, M. Z. Wang, Y. G. Yang, R. Cai and D. Sheng Su, Chem. Commun., 2012, 48, 7149 RSC.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020 CrossRef CAS PubMed.
- Y. Jiao, D. Han, L. Liu, L. Ji, G. Guo, J. Hu, D. Yang and A. Dong, Angew. Chem., Int. Ed., 2015, 54, 5727 CrossRef CAS PubMed.
- Y. Jiao, D. Han, Y. Ding, X. Zhang, G. Guo, J. Hu, D. Yang and A. Dong, Nat. Commun., 2015, 6, 6420 CrossRef PubMed.
- X. Wang, Y. Zhang, C. Zhi, X. Wang, D. Tang, Y. Xu, Q. Weng, X. Jiang, M. Mitome and D. Golberg, Nat. Commun., 2013, 4, 2905 Search PubMed.
- L. Qiu, J. Z. Liu, S. L. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- H. Wang, K. Sun, F. Tao, D. J. Stacchiola and Y. H. Hu, Angew. Chem., Int. Ed., 2013, 52, 9210 CrossRef CAS PubMed.
- J. S. Lee, H. J. Ahn, J. C. Yoon and J. H. Jang, Phys. Chem. Chem. Phys., 2012, 14, 7938 RSC.
- H. S. Ahn, J. W. Jang, M. Seol, J. M. Kim, D. J. Yun, C. Park, H. Kim, D. H. Youn, J. Y. Kim, G. Park, S. C. Park, J. M. Kim, D. I. Yu, K. Yong, M. H. Kim and J. S. Lee, Sci. Rep., 2013, 3, 1396 Search PubMed.
- J. Hu, Z. Kang, F. Li and X. Huang, Carbon, 2014, 67, 221 CrossRef CAS PubMed.
- Z. Yang, C. Y. Chen, C. W. Liu and H. T. Chang, Chem. Commun., 2010, 46, 5485 RSC.
- J. G. Radich, R. Dwyer, P. V. Kamat and J. Phys, Chem. Lett., 2011, 2, 2453 CAS.
- F. Gong, H. Wang, X. Xu, G. Zhou and Z. S. Wang, J. Am. Chem. Soc., 2012, 134, 10953 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07957h |
| This journal is © The Royal Society of Chemistry 2015 |