
Enhanced amoxicillin treatment using the electro-peroxone process: key factors and degradation mechanism
Wanqian Guo*,
Qu-Li Wu,
Xian-Jiao Zhou,
Hai-Ou Cao,
Juan-Shan Du,
Ren-Li Yin and
Nan-Qi Ren
State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, 73 Huanghe Road, Harbin, Heilongjiang 150090, P. R. China
First published on 9th June 2015
Abstract
Amoxicillin (AMO) degradation was investigated using electrolysis, ozonation, and the electro-peroxone (E-peroxone) process. The E-peroxone process was found to be the most effective for AMO degradation. 67.8% total organic carbon (TOC) mineralization was obtained after 60 min by the E-peroxone process. In comparison, only 47.3% and 3.1% TOC mineralization were obtained using individual ozonation and electrolysis processes, respectively. It was found that hydroxyl radical production and O3 utilization were both enhanced in the E-peroxone process. The effect of pH on the E-peroxone process was investigated, and the highest AMO removal rate was obtained at pH = 9, indicating pH control was crucial in the E-peroxone process. In addition, more oxidation typical intermediates were identified in the E-peroxone process than the ozonation process using UPLC-MS/MS. Different pathways of AMO degradation were proposed, involving the hydroxylation of the benzoic ring and N, the four-membered β-lactamic ring opening, the oxidation of S, and other bond cleavage reactions. All these results above indicated that the introduction of electrolysis in ozonation has enhanced AMO cleavage and hence its degradation.
Introduction
Antibiotics are widely used for curing human and veterinary diseases, and are also used as feed additives for livestock growth. Due to the potential adverse effects on aquatic ecology1 and human health,2–4 the presence of antibiotics in aquatic environments received special concern. Recent studies reported that the antibiotics concentration in aquatic environments ranged from ng L−1 to μg L−1 in USA, Canada, Germany and China.5–8 Amoxicillin (AMO), a broad-spectrum amino penicillin antibiotic, is widely used as a kind of veterinary antibiotic in aquaculture, animal husbandry and also human medicine. Terribly, AMO has been found in a certain amount in water environments that threatens human health. It was reported the AMO concentration was up to about 0.6 μg L−1 in British rivers,9 as well as 13 ng L−1 in a municipal sewage treatment plant in Italy.10 Due to its antibacterial nature and its toxicity, AMO shows resistance to conventional biological water treatment methods. Thus, it is necessary to develop efficient treatment techniques to prevent AMO entering into aquatic environments.Ozonation was proposed to be a suitable process for antibiotics treatment (removal), due to the strong oxidation ability of ozone (E0 = 2.07 V). Ozone molecular was proved to have a high selectivity in attacking conjugated double bonds (e.g., N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CN, and CC), aromatic bonds or nitrogen, phosphorous, oxygen or sulphur atoms,11 since it only selectively reacted with nucleophilic molecules.12 Previous investigations have already demonstrated that ozone is capable of attacking those present β-lactams antibiotics (including AMO) in water. Although high removal rates were achieved, the degree of mineralization was low (<20%), even undergoing for a long treatment time.13,14 Due to the low mineralization degree, biodegradability enhancement (increment of BOD5/COD ratio) was slight after the ozonation treatment.15
N, CN, and CC), aromatic bonds or nitrogen, phosphorous, oxygen or sulphur atoms,11 since it only selectively reacted with nucleophilic molecules.12 Previous investigations have already demonstrated that ozone is capable of attacking those present β-lactams antibiotics (including AMO) in water. Although high removal rates were achieved, the degree of mineralization was low (<20%), even undergoing for a long treatment time.13,14 Due to the low mineralization degree, biodegradability enhancement (increment of BOD5/COD ratio) was slight after the ozonation treatment.15
To improve the mineralization efficiency, combined ozone processes were used in antibiotic wastewater treatment, particularly combined O3 and electrolysis process (the so-called electro-peroxone process). The main mechanism of electro-peroxone is that O3 can react with H2O2 which is generated by the electrical process in situ, to form hydroxyl radical (˙OH),16,17 which can improve the mineralization efficiencies of pollutants than ozonation remarkably.18 In addition, electro-peroxone process produced none secondary pollutants, only leaving H2O and O2 as by-products.19,20 Therefore, electro-peroxone process was considered as an effective and environmental-friendly advanced oxidation technology for wastewater treatment.18,21 Meanwhile, electro-peroxone has some advantages over peroxone process such as that the addition of small amounts of hydrogen peroxide increased the removal efficiency (up to 15%) and the effluents biodegradability, the biotoxicity was not removed completely.22,23 However, H2O2 is unsafe to transport, store and handle, due to its high reactivity. High concentrations of H2O2 addition would decrease the process efficiency, since excess H2O2 may act as a free radical scavenger. Electro-peroxone process would use the H2O2 more economically and efficiently than other individual process. Particularly, E-peroxone processes have already been applied to the dye wastewater treatment and landfill leachate treatment successfully.24,25 Despite so many advantages, the AMO wastewater treatment using E-peroxone has not been reported yet.
This study focused on the performance of the E-peroxone process by using activated carbon fiber (ACF) cathode for antibiotic removal, and chose AMO as the model compound. The main objective of this work was to examine the feasibility of AMO treatment using E-peroxone. The operation and AMO degradation pathways were also discussed.
Experimental
Chemicals and reagents
AMO (C16H18N3NaO5S, analytical reagent, 99.0%) was purchased from TCI and used as received without further purification. Hydroxyterephthalic acid (HTA, 97%) was purchased from Sigma-Aldrich. Terephthalic acid (TA, >99%) was purchased from Alfa Aesar. Other chemicals (e.g. Na2SO4, NaOH, and K2HPO4, H3PO4 and KH2PO4) were analytical grade and purchased from Tianjin chemical Works Co., China. All solutions were prepared using deionized water.Ozonation, electrolysis, and E-peroxone process of AMO
AMO solutions were buffered by the addition of K2HPO4, H3PO4 and KH2PO4. Oxidation experiments were carried out in a 1.0 L reactor with a continuous supply of O3. Ozonation, electrolysis, and E-peroxone process of 1.0 L AMO (initial concentration of 100 mg L−1) were carried out in an undivided acrylic cube reactor designed originally. For ozonation process, ozone was produced from pure O2 gas (99.9%) using an ozone generator with gaseous flow meter (DHX-SS-1G, Jiujiu ozone, Harbin, China). The ozone effluent was placed on the bottom of the reactor, using a fine bubble diffuser at a constant flow rate of 0.4 L min−1 unless otherwise specified. The gaseous product from the reactor was led to a terminator, where the remaining ozone was absorbed by KI solution. Both the electrolysis and E-peroxone processes were conducted under galvanostatic conditions using a DC power supply (DJS-292, Leici Co., Shanghai, China). The anode was a 6 cm2 plate (2 cm × 3 cm) made of Pt, while the cathode was a 48 cm2 (6 cm × 8 cm) activated carbon fiber electrode. The supporting electrolyte was Na2SO4 solution 0.05 M. The electrolysis only process was initiated by turning on the DC power supply while the ozone generator was off. So-called E-peroxone process meant the DC power supply was turned on after the ozone generator was switched on for 15 min to reach an almost steady ozone concentration. The mixture of O2 and O3 from the ozone generator, was bubbled into the reactor at 0.4 L min−1, which is the same flow rate as in ozonation process. The ozonation, electrolysis, and E-peroxone process were run for 60 min. The runs were carried out at pH 3, 5, 7, 9 and 11, ozone dose was 3 g h−1 and applied current density was 400 mA unless otherwise specified. During ozonation, electrolysis, and E-peroxone process of AMO treatment, an aliquot of solution sample was collected from the reactor at various time intervals. Each run in the research was performed twice to ensure reproducibility.Analytical methods
The concentration of AMO was determined by HPLC (Waters, C18, λ = 254 nm). The mobile phase was a mixture of acetonitrile and 0.1% acetic acid with the volume ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95. A 10 μL volume was injected using the auto sampler. TOC was measured using a TOC-VCPH analyzer (Shimadzu Co. Japan) to evaluate the mineralization of AMO during the treatment process.
:95. A 10 μL volume was injected using the auto sampler. TOC was measured using a TOC-VCPH analyzer (Shimadzu Co. Japan) to evaluate the mineralization of AMO during the treatment process.
The concentration of O3 in solution was measured using the indigo method.26 The ˙OH concentration was analyzed using a terephthalic acid (TA) trapping protocol. Briefly, the initial concentrations of TA (2 mM) and NaOH (5 mM) were added into the electrolyte in the reactor before the E-peroxone process started. And then the E-peroxone system was turned on, to generate ˙OH in the electrolyte. The ˙OH generated continuously during the E-peroxone process, then was trapped by the TA which is non-fluorescent, to form HTA which is highly fluorescent. The HTA concentration was determined using a fluorescence spectrophotometer (Hitachi, F-7000), which can be taken as a cumulative measurement of the ˙OH produced during the operation time.27,28
The intermediates of AMO degradation were measured by UPLC-MS/MS system, which consisted of a Waters ACQUITY UPLC instrument coupled to a TQD triple-quadrupole mass spectrometer (Waters Corp., Milford, MA). Separations were performed on an ACQUITY UPLC BEH C18 column (100 mm × 2.1 mm) with a 1.7 μm particle size equipped with a 0.2 μm pre-column filter unit and a guard column (Waters Corp.). The flow rate was set at 0.1 mL min−1. The column and autosampler tray temperature were both set at 40 °C. The MS/MS instrument was operated with a capillary voltage of 1.00 kV, a source temperature of 350 °C and desolvation gas (nitrogen) at 350 °C with a flow of 900 L h−1. The interchannel delay was 20 ms. Parent and daughter ions, cone voltage and collision energy were optimized by automatic infusion of 1 mg L−1 in a mixture of 50/50 water/acetonitrile containing 0.1% formic acid. Analysis was measured in positive electrospray ionisation (ESI+) mode; the mobile phase consisted of a mixture of solution A (0.1% formic acid in water) and solution B (0.1% formic acid in acetonitrile) with an initial composition of 90% solution A and 10% solution B. The mobile phase composition changed linearly from 10% solution B to 40% at 10.0 min, then solution B was re-equilibrated to starting conditions in 0.5 min and maintained for 1.5 min.29
Results and discussion
Comparison of different process for AMO removal
Only 37% AMO was removed after 6 min treatment by electrolysis. While, AMO was completely degraded within 5 min and 4 min by ozonation process and E-peroxone process, respectively (Fig. 1a). The E-peroxone process enhanced the degradation rate of AMO comparing to the other two individual processes. The low removal rate of AMO in electrolysis resulted from the limited mass transfer rate of AMO molecules to the anode surface and the limited oxidation capacity of H2O2.19,30–32 The ozonation process degraded the AMO slower than E-peroxone, indicating that O3 utilization and ˙OH production were somewhat different in ozonation and E-peroxone process and influenced the degradation rate.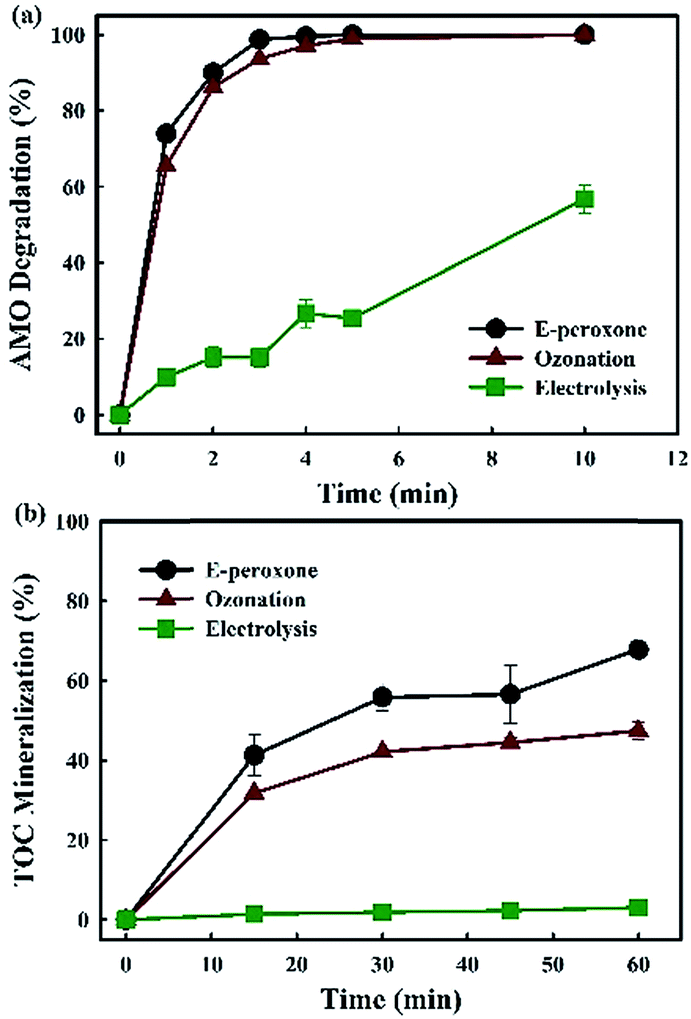 | ||
| Fig. 1 Degradation of AMO wastewater and TOC mineralization by electrolysis, ozonation and E-peroxone treatment (current of 400 mA; inlet O3 concentration of 4 g h−1). | ||
In addition, mineralization of AMO by different process was also evaluated in this work. Only 3.1% and 47.3% TOC mineralization was achieved after 60 min in electrolysis and ozonation process, respectively (Fig. 1b). When combined these two techniques (ozonation and electrolysis) together, so-called E-peroxone process, more than 67.8% TOC was removed in the same reaction time (Fig. 1b). Similarly with AMO degradation, low mineralization in electrolysis alone process resulted from the mass transfer limitation of AMO molecules to the anode surface as described for the low degradation degree.19,30 As for ozonation, although O3 could completely destruct the AMO structure in 5 min, it reacted rarely with the aliphatic carboxylic acids formed from the oxidation intermediates.33,34 In the long time mineralization of AMO, the ˙OH production in E-peroxone seemed to play an important role.
It was shown that E-peroxone process provided a feasible and promising way to remove such kind of antibiotics and their intermediate products. However, how electricity introduction enhanced the O3 utilization and ˙OH production in ozonation process for AMO treatment deserved further research.
˙OH production and aqueous O3 concentration in E-peroxone process
To understand the mechanism and evaluate the effect of electricity introduction in E-peroxone process, aqueous O3 concentration and ˙OH production were compared in these two processes. The measurement of ˙OH production in the solution was through HTA production, which was the product by “˙OH and TA” reaction.Fig. 2 showed that when pure O2 was sparged into the reactor during electrolysis (while the ozone generator was off), the H2O2 concentration increased almost linearly with reaction time. The result indicated that H2O2 was continuously produced from the sparged O2 at the ACF cathode, which was consistent with eqn (1). Conversely, when the ozone generator was turned on, the O2 and O3 gas mixture was sparged into the reactor, no H2O2 accumulation was observed.
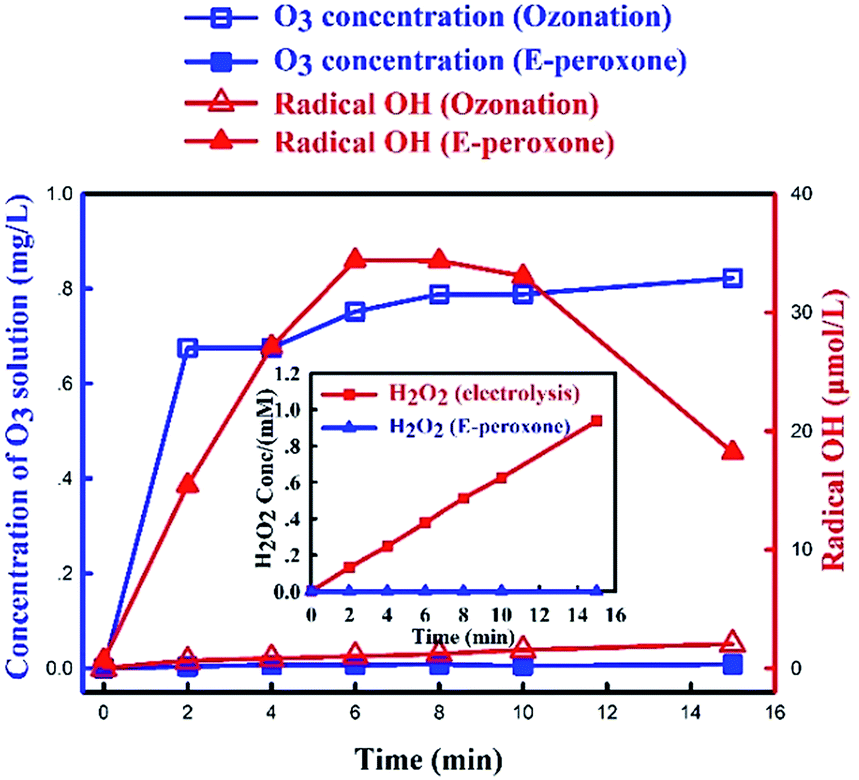 | ||
| Fig. 2 Concentration of O3 solution and radical OH during ozonation, and E-peroxone process and H2O2 concentration during electrolysis and E-peroxone in ionized water (current of 400 mA; inlet O3 concentration of 4 g h−1). | ||
In E-peroxone process, the concentration of HTA increased significantly within the first 8 min (Fig. 2). The result indicated that in E-peroxone process the sparged O3 and in situ generated H2O2 reacted actively to continuously produce ˙OH (eqn (2)), and then the produced ˙OH and TA formed HTA. After 8 min reaction time, HTA concentration decreased, since most TA had reacted with ˙OH, and then ˙OH and O3 would consume HTA gradually.28 In contrast, HTA was substantially low throughout the whole ozonation process. These results demonstrated that dissolved O3 was consumed in the reaction with electro-generated H2O2 and electrochemical reactions such as cathode reduction to ˙OH in the E-peroxone process.17,24 As a result, ˙OH production was significantly enhanced in E-peroxone process, which contributed to higher AMO degradation rate in E-peroxone comparing to in ozonation process.
| O2 + 2H+ + 2e− → H2O2 | (1) |
| O3 + H2O2 + e− → ˙OH + O2 + ˙OH− | (2) |
In ozonation process, the aqueous phase O3 concentration increased rapidly to a plateau at ∼0.8 mg L−1. In comparison, the aqueous O3 concentration was substantially low during the whole E-peroxone treatment. These results indicated that dissolved O3 was rapidly consumed in E-peroxone process. According to mass transfer theories,35 these electrochemically-driven reactions would enhance O3 mass transfer from the gas phase to the liquid phase.
Consequently, the effective ozone dose would be higher in E-peroxone process than in ozonation. In other word, more sparged O3 was transferred to the liquid phase for pollutants degradation in E-peroxone process rather than running out into gas phase in ozonation process.
The above results showed that in E-peroxone process, considerable amounts of ˙OH could be produced, and higher utilization rate of O3 was obtained as well. Therefore, it could be a reasonable interpretation for E-peroxone process performed more effective and economical than ozonation process for AMO degradation.
Effects of operating parameters on AMO degradation and TOC mineralization in E-peroxone
In order to control the E-peroxone treatment of amoxicillin process efficiently and economically, important operating parameters such as O3 concentration, current, and solution pH on the process performance were evaluated systematically.Fig. 3 showed that increasing the O3 concentration in the sparged gas enhanced both amoxicillin degradation and TOC mineralization in the E-peroxone process. This result can be easily rationalized because increasing the gas phase O3 concentration would enhance the mass transfer of O3 from gas phase to liquid phase, which leads to higher ˙OH production rates from the reaction of aqueous O3 with the in situ generated H2O2 in the solution. Consequently, amoxicillin and its degradation intermediates can be more rapidly mineralized as the gas phase O3 concentration is increased.
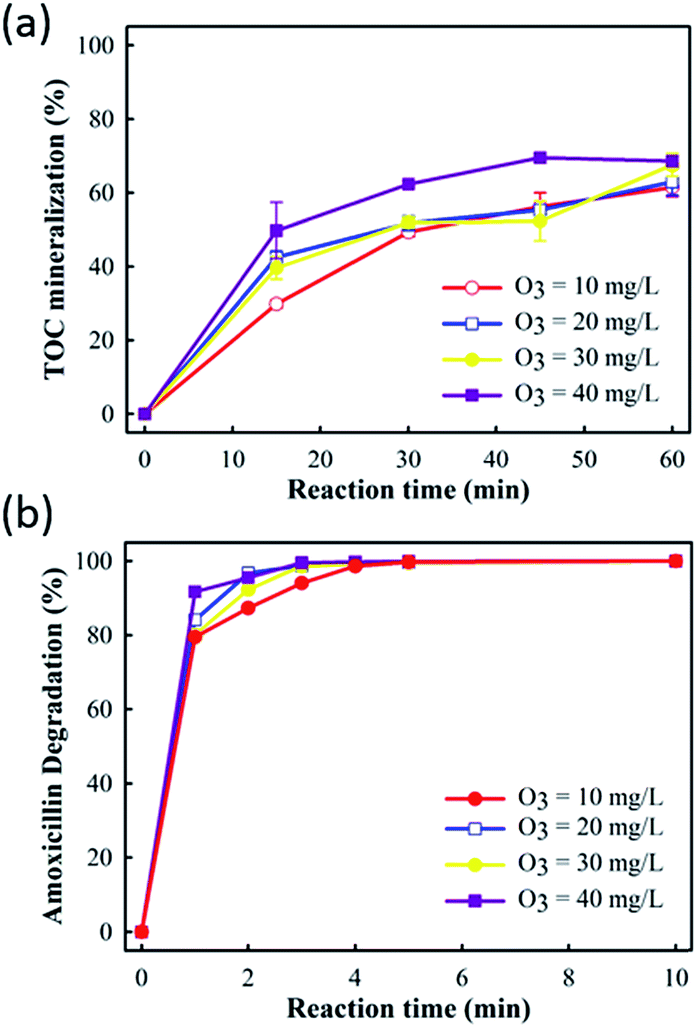 | ||
| Fig. 3 Effects of O3 concentration on AMO degradation and TOC mineralization in E-peroxone process (current of 400 mA). | ||
Fig. 4 showed that amoxicillin degradation and TOC mineralization increased as the applied current increased from 100 to 300 mA. However, further increasing the current to 400 mA did not enhance amoxicillin degradation and TOC mineralization accordingly. Consequently, increasing the applied current can produce more aqueous ˙OH when sufficient aqueous O3 is available to react with the electro-generated H2O2, leading to enhanced pollutant degradation by the E-peroxone process. However, due to the low solubility of O3, the ˙OH production rate would eventually be limited by the rate of O3 transfer from gas phase to liquid phase when the current is increased beyond a critical value. When the amount of aqueous O3 is insufficient in the solution, the excess H2O2 will contribute little to TOC mineralization because it is not a powerful oxidant. Therefore, the increasing the current beyond 300 mA did not increase the rate of amoxicillin degradation and TOC mineralization further, in the E-peroxone process.
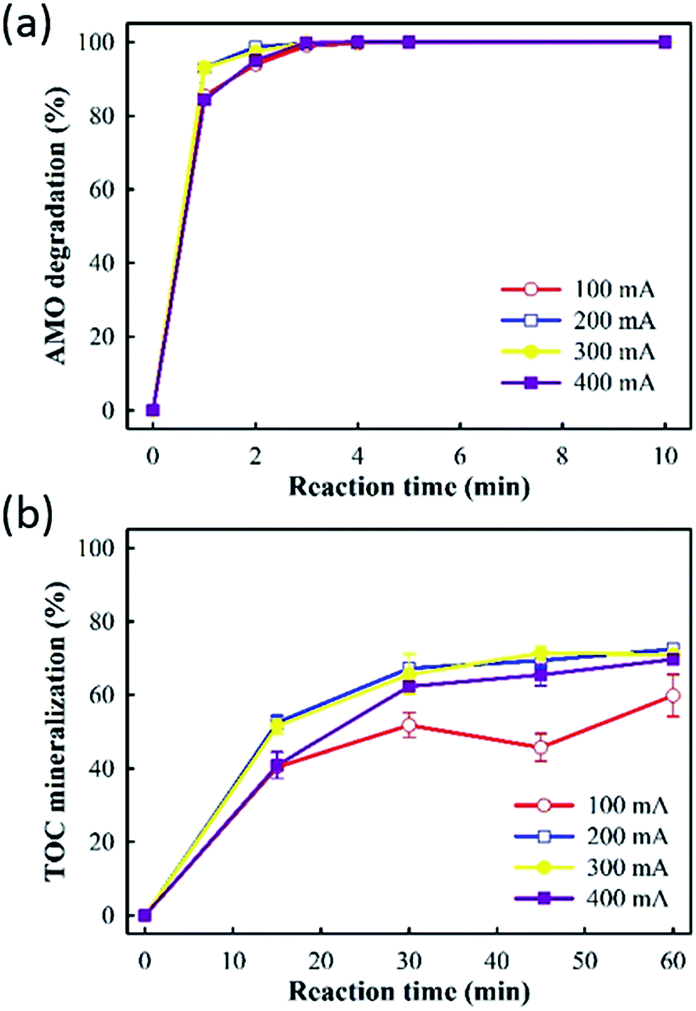 | ||
| Fig. 4 Effects of current on AMO degradation and TOC mineralization in E-peroxone process (inlet O3 concentration of 4 g h−1). | ||
In traditional ozonation treatment, pH is an important parameter, as ozone oxidation pathways include direct oxidation by molecular ozone under acidic conditions while indirect oxidation by ˙OH under alkaline pH values. However, how pH affected oxidation type and AMO structure present in E-peroxone process were both unclear. In the present study, effects of pH on AMO treatment in E-peroxone process were designed to clarify.
pH is usually considered as an important parameter for hydroxide ions initiate ozone decomposition, which involves the following reactions:36
| O3 + OH− → HO2− + O2; k = 70 M−1 s−1; | (3) |
| O3 + HO2− → ˙OH + O2− + O2; k = 2.8 × 106 M−1 s−1; | (4) |
| HO2− + ˙OH → ˙O2H + ˙OH−; | (5) |
In E-peroxone process, when pH value was lower than 7.0, AMO degradation and TOC mineralization rate increased with pH (Fig. 5a and b). However, the enhancement effect of pH on AMO removal was limited when its value exceeded 7.0, and the highest removal rate was obtained at pH = 9, while the degradation and TOC mineralization rates of AMO decreased considerably when pH = 11. The result indicated that the effect of pH on AMO degradation was complicated and it varied at different solution pHs. For better understanding, the mechanism of these limited factors were discussed in detail.
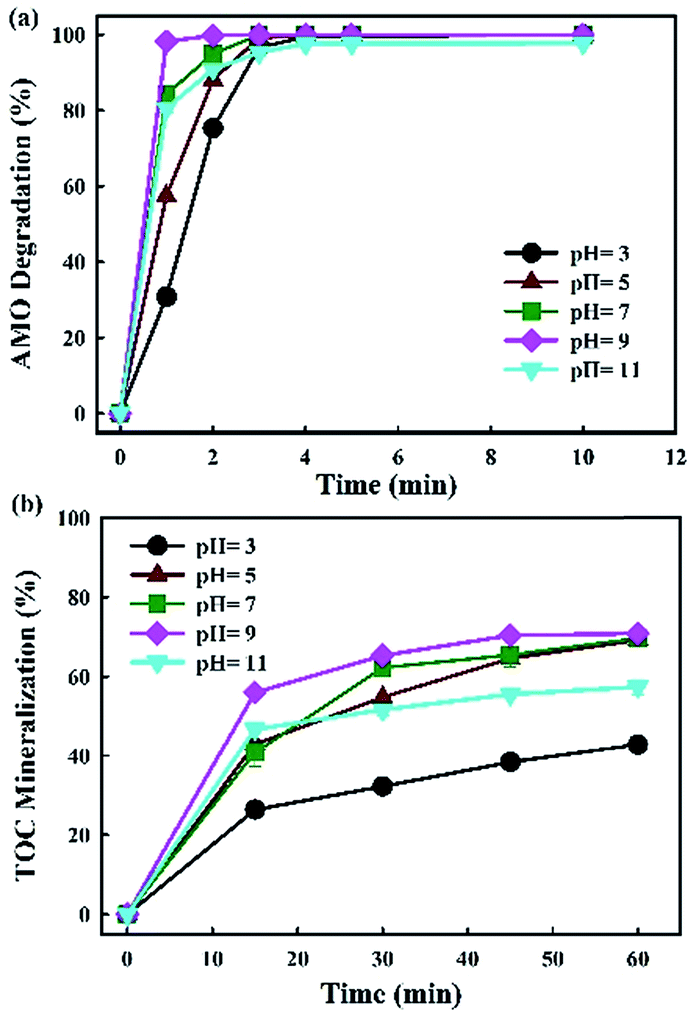 | ||
| Fig. 5 Effects of initial pH on AMO degradation and TOC mineralization in E-peroxone process (current of 400 mA; inlet O3 concentration of 4 g h−1). | ||
Under acidic condition, the dissociation of H2O2 to HO2− would increase with pH. Further, H2O2 reacted with O3 only when present as the anion, HO2−. The increase of H2O2 thus enhanced ˙OH generation in E-peroxone process via eqn (4). This reaction well explained why AMO degradation and TOC mineralization rate went faster with the increase of pH.
Under alkali condition, it was accepted that aqueous ozone decomposing to HO2− (eqn (3)) could enhance ˙OH generation in conventional ozonation process.36 However, it might occur a side reaction and decrease the degradation efficiency when pH was at 11 in this study. It was shown that the availability of aqueous O3 was substantially low when the E-peroxone process was conducted at 400 mA and neutral pH (Fig. 5). Increasing pH to 11 had further decreased the availability of aqueous O3, due to the decomposition of O3, causing the decrease in ˙OH production (eqn (4)). Meanwhile, the dissociation of H2O2 would be enhanced with pH increase in E-peroxone process, as the pKa of H2O2 was 11.6 at which favoured the production of excessive HO2− and further acted as a scavenger of ˙OH via eqn (5). These reactions may account for the decrease in AMO degradation efficiency and lower TOC mineralization in E-peroxone process when E-peroxone (400 mA) was conducted at pH 11 than pH 7.
The solution pH not only affected the decomposition of aqueous O3, but also influenced the existing forms of AMO, which affected AMO degradation efficiency in turn. AMO has three pKa values of 2.68, 7.49 and 9.63 resulting in protonated, non-protonated and deprotonated forms, which could influence its degradation efficiency.37 Considering the structure of the AMO, the pKa1 value of AMO carboxylic group is 2.68, and the protonation of carboxylic group formed at pH 2.6. Decreasing solution pH from 7 to 2.6 enhanced protonation of carboxylic acid products formed from AMO degradation. The protonation of AMO and carboxylic intermediates would generally decrease their susceptibility to ˙OH oxidation.38 This could explain why E-peroxone process was more effective when it was operated at pH 7 than at pH 2.6, for AMO degradation and TOC mineralization. These results above showed that pH played an important role in AMO degradation, and pH controlled at 7 to 9 could guarantee hydroxyl radical production and AMO susceptibility to ˙OH oxidation in E-peroxone process.
Intermediates and AMO degradation pathway by ozonation and E-peroxone process
Although it seemed that E-peroxone process and ozonation had achieved similar removal effects on AMO, E-peroxone process had its own priority in AMO mineralization. Thus, it was assumed that the electricity influenced the AMO degradation on the intermediates species and pathways.To investigate the influence of electricity on AMO degradation, the intermediates were identified precisely by UPLC-ESI-MS-MS. The degradation by-products were determined for AMO by ozonation and E-peroxone process corresponding with the different treatment process at starting pH of 6.0. 10 intermediates were detected in ozone process, while 15 intermediates were detected in E-peroxone process (Table 1). The main fragments observed in the mass spectra of each intermediate are indicated on Fig. 6 and 7.
| Compound (m/z) | Structure | Ozonation | E-peroxone |
|---|---|---|---|
| a D = detected fragmentation, ND = not detected fragmentation. | |||
| 139 | 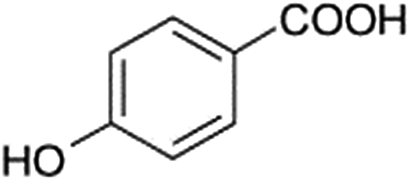 |
D | D |
| 160 | 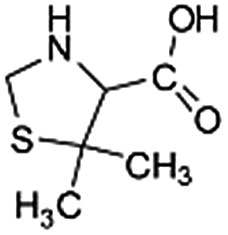 |
ND | D |
| 176 | 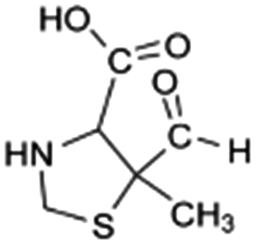 |
D | D |
| 231 | 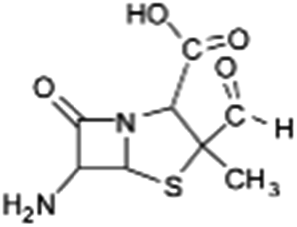 |
ND | D |
| 239 | 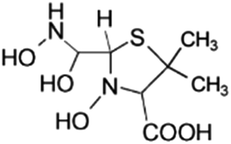 |
ND | D |
| 310 | 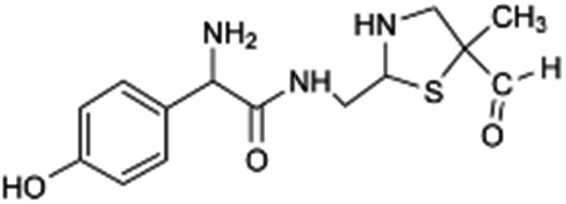 |
ND | D |
| 340 | 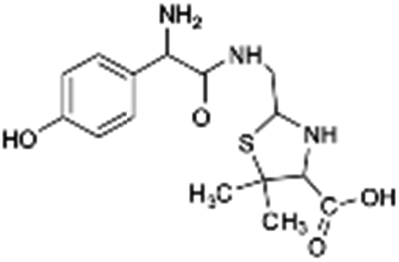 |
ND | D |
| 354 | 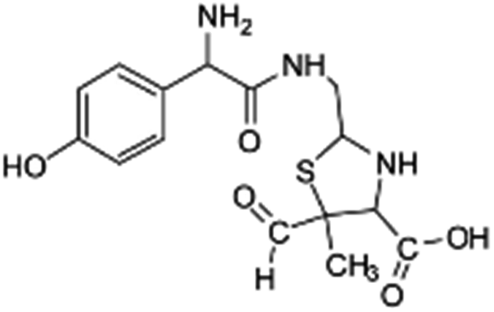 |
D | D |
| 377 | 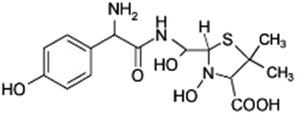 |
ND | D |
| 382 | 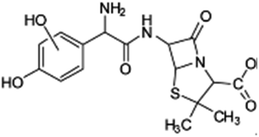 |
D | D |
| 383 | 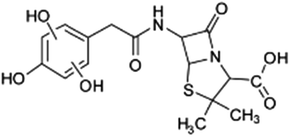 |
D | D |
| 384 | 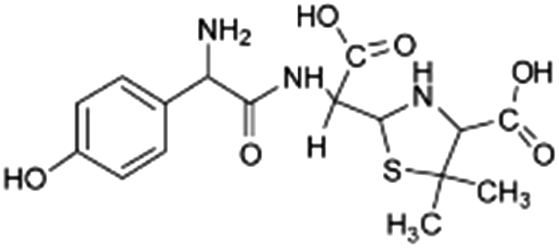 |
D | D |
| 398 | 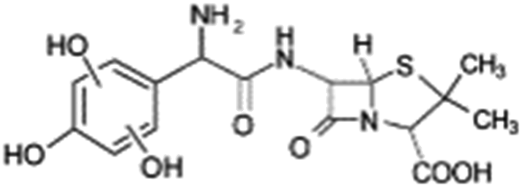 |
D | D |
| 400 | 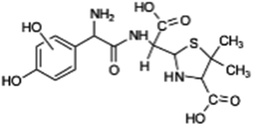 |
D | D |
| 412 | 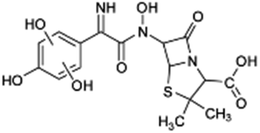 |
D | D |
| 428 | 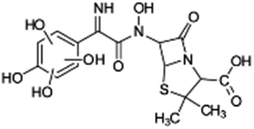 |
D | D |
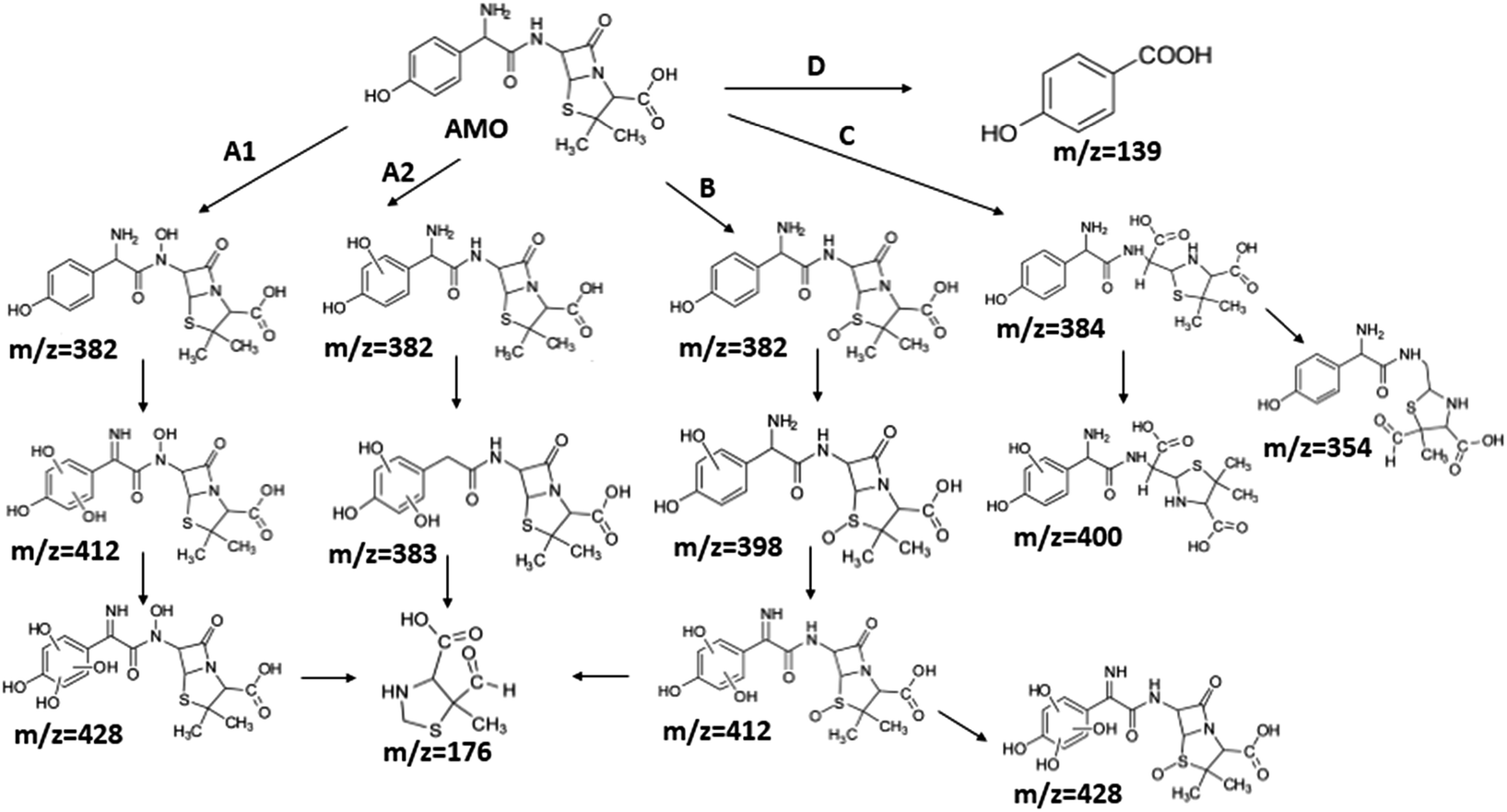 | ||
| Fig. 6 Proposed degradation pathway of AMO by ozonation. | ||
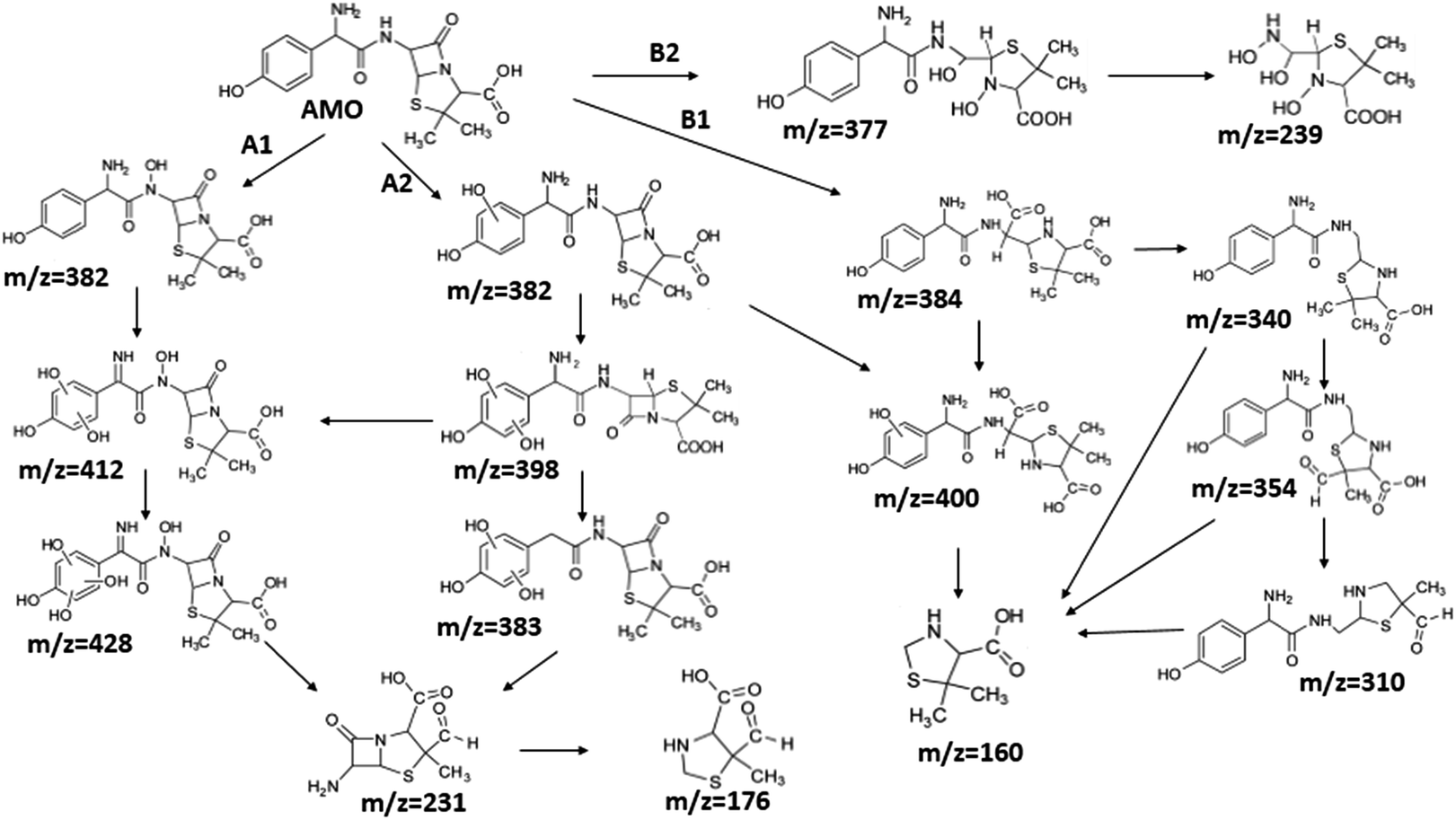 | ||
| Fig. 7 Proposed degradation pathway of AMO by E-peroxone. | ||
The bond cleavage generated at compounds (m/z = 383, 412 and 428) and further degradation losing the CO group and the four-membered β-lactam ring was evidenced compound m/z = 176.
Another degradation pathway (Fig. 7B1) corresponded to the opening of the four-membered β-lactam ring and yielded the penicilloic acid (m/z = 384) and a series of derivatives (m/z = 340, 354, 310 and 400). A further decarboxylation reaction yielded the intermediates m/z = 340. The oxidation of the methyl groups in the thiazolidine ring was evidenced by the identification of the intermediates m/z = 354 and m/z = 310. The product with m/z = 160 could be also generated by the cleavage of the former derivatives.13 The other pathway (Fig. 7B2) began with the destruction of lactamic bond yielding the intermediates (m/z = 377) with its subsequent degradation to a product with m/z = 239.40
The bond cleavage between nitrogen of the amino group and the carbonyl group was evidenced by compound m/z = 231 and further degradation, due to the loss of the CO group and of the four-membered β-lactam ring, yielding the intermediates m/z = 176.
It was found the introduction of electrolysis in ozonation had enhanced the cleavage of AMO, and then degraded to smaller products, so that AMO became easier to be attacked. Consequently, the removal rate of TOC was increased in E-peroxone process, comparing to ozone-alone process.
Conclusions
High removal efficiency and mineralization rate was obtained by E-peroxone over ozonation and electrolysis synergistic effect for amoxicillin treatment, owing to more effective hydroxyl radical production introduced by E-peroxone process. AMO was completely degraded in 4 min, and 67.8% total organic carbon (TOC) mineralization was obtained after 60 min by E-peroxone proecess. It was found that pH played an important role in AMO degradation and TOC mineralization, which not only affecting the decomposition of aqueous O3 and the hydroxyl radical production, but also affecting the existing forms of AMO. The highest removal rates were obtained at pH = 9, indicating pH control was crucial in E-peroxone process.The degradation pathways were deduced in two processes, based on the reaction between AMO and O3 or ˙OH. 15 intermediates were identified in E-peroxone process while 10 intermediates were detected in ozone process using UPLC-MS/MS. These intermediates were generated in the following steps, the hydroxylation of the benzoic ring and N, the four-membered β-lactamic ring opening, oxidation of S, and other bond cleavage reactions. The introduction of electrolysis in ozonation had improved AMO degradation and increased TOC removal, suggesting E-peroxone process is feasible and has great potential for enhancing AMO treatment.
Acknowledgements
This work was financially supported by National Nature Science Foundation of China (51121062). The authors also gratefully acknowledge the financial support by State Key Laboratory of Urban Water Resource and Environment (2014TS06), National science and technology plan of China (2014BAD02B03), and Fund for young top-notch talent teachers by Harbin Institute of Technology (AUGA5710052514).Notes and references
- L.-H. Yang, G.-G. Ying, H.-C. Su, J. L. Stauber, M. S. Adams and M. T. Binet, Environ. Toxicol. Chem., 2008, 27, 1201–1208 CrossRef CAS PubMed
.
- S. K. Khetan and T. J. Collins, Chem. Rev., 2007, 107, 2319–2364 CrossRef CAS PubMed
- J.-P. Besse and J. Garric, Toxicol. Lett., 2008, 176, 104–123 CrossRef CAS PubMed
- F. Pomati, S. Castiglioni, E. Zuccato, R. Fanelli, D. Vigetti, C. Rossetti and D. Calamari, Environ. Sci. Technol., 2006, 40, 2442–2447 CrossRef CAS
- K. Kuemmerer, Chemosphere, 2009, 75, 417–434 CrossRef CAS PubMed
- K. Kuemmerer, Chemosphere, 2009, 75, 435–441 CrossRef CAS PubMed
- P. Verlicchi, M. Al Aukidy and E. Zambello, Sci. Total Environ., 2012, 429, 123–155 CrossRef CAS PubMed
- H. W. Leung, T. B. Minh, M. B. Murphy, J. C. W. Lam, M. K. So, M. Martin, P. K. S. Lam and B. J. Richardson, Environ. Int., 2012, 42, 1–9 CrossRef CAS PubMed
- B. Kasprzyk-Hordern, R. M. Dinsdale and A. J. Guwy, Water Res., 2008, 42, 3498–3518 CrossRef CAS PubMed
- S. Castiglioni, R. Bagnati, R. Fanelli, F. Pomati, D. Calamari and E. Zuccato, Environ. Sci. Technol., 2006, 40, 357–363 CrossRef CAS
- K. Ikehata, N. J. Naghashkar and M. G. Ei-Din, Ozone: Sci. Eng., 2006, 28, 353–414 CrossRef CAS PubMed
- H. Stockinger, E. Heinzle and O. M. Kut, Environ. Sci. Technol., 1995, 29, 2016–2022 CrossRef CAS PubMed
- R. Andreozzi, M. Canterino, R. Marotta and N. Paxeus, J. Hazard. Mater., 2005, 122, 243–250 CrossRef CAS PubMed
- I. A. Balcioglu and M. Otker, Chemosphere, 2003, 50, 85–95 CrossRef
- R. F. Dantas, S. Contreras, C. Sans and S. Esplugas, J. Hazard. Mater., 2008, 150, 790–794 CrossRef CAS PubMed
- J. Staehelin and J. Hoigne, Environ. Sci. Technol., 1982, 16, 676–681 CrossRef CAS
- S. Yuan, Z. Li and Y. Wang, Electrochem. Commun., 2013, 29, 48–51 CrossRef CAS PubMed
- J. Pablo Pocostales, M. M. Sein, W. Knolle, C. von Sonntag and T. C. Schmidt, Environ. Sci. Technol., 2010, 44, 8248–8253 CrossRef PubMed
- C. A. Martinez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef CAS PubMed
- A. Xu, X. Li, S. Ye, G. Yin and Q. Zeng, Appl. Catal., B, 2011, 102, 37–43 CrossRef CAS PubMed
- R. G. Rice, Ozone: Sci. Eng., 1997, 18, 477–515 CrossRef CAS PubMed
- I. Arslan-Alaton and A. E. Caglayan, Ecotoxicol. Environ. Saf., 2006, 63, 131–140 CrossRef CAS PubMed
- B. De Witte, J. Dewulf, K. Demeestere and H. Van Langenhove, J. Hazard. Mater., 2009, 161, 701–708 CrossRef CAS PubMed
- B. Bakheet, S. Yuan, Z. Li, H. Wang, J. Zuo, S. Komarneni and Y. Wang, Water Res., 2013, 47, 6234–6243 CrossRef CAS PubMed
- Z. Li, S. Yuan, C. Qiu, Y. Wang, X. Pan, J. Wang, C. Wang and J. Zuo, Electrochim. Acta, 2013, 102, 174–182 CrossRef CAS PubMed
- H. Bader and J. Hoigne, Water Res., 1981, 15, 449–456 CrossRef CAS
- N. Milan-Segovia, Y. Wang, F. S. Cannon, R. C. Voigt and J. C. Furness Jr, Ozone: Sci. Eng., 2007, 29, 461–471 CrossRef CAS PubMed
- I. Hua and M. R. Hoffmann, Environ. Sci. Technol., 1997, 31, 2237–2243 CrossRef CAS
- M. Carlier, V. Stove, J. A. Roberts, E. Van de Velde, J. J. De Waele and A. G. Verstraete, Int. J. Antimicrob. Agents, 2012, 40, 416–422 CrossRef CAS PubMed
- E. Brillas, I. Sires, C. Arias, P. L. Cabot, F. Centellas, R. M. Rodriguez and J. A. Garrido, Chemosphere, 2005, 58, 399–406 CrossRef CAS PubMed
- A. Oezcan, M. A. Oturan, N. Oturan and Y. Sahin, J. Hazard. Mater., 2009, 163, 1213–1220 CrossRef CAS PubMed
- J. Hastie, D. Bejan, M. Teutli-Leon and N. J. Bunce, Ind. Eng. Chem. Res., 2006, 45, 4898–4904 CrossRef CAS
- A. Lopez, H. Benbelkacem, J. S. Pic and H. Debellefontaine, Environ. Technol., 2004, 25, 311–321 CrossRef CAS PubMed
- M. J. Quero-Pastor, M. C. Garrido-Perez, A. Acevedo and J. M. Quiroga, Sci. Total Environ., 2014, 466, 957–964 CrossRef PubMed
- E. L. Cussler, Diffusion: mass transfer in fluid systems, Cambridge university press, 2009 Search PubMed
- U. von Gunten, Water Res., 2003, 37, 1443–1467 CrossRef CAS
- A. F. Goddard, M. J. Jessa, D. A. Barrett, P. N. Shaw, J. P. Idstrom, C. Cederberg and R. C. Spiller, Gastroenterology, 1996, 111, 358–367 CrossRef CAS PubMed
- G. V. Buxton, C. L. Greenstock, W. P. Helman, A. B. Ross and W. Tsang, J. Phys. Chem. Ref. Data, 1988, 17, 513 CrossRef CAS PubMed
- A. G. Trovo, R. F. P. Nogueira, A. Aguera, A. R. Fernandez-Alba and S. Malato, Water Res., 2011, 45, 1394–1402 CrossRef CAS PubMed
- D. Klauson, J. Babkina, K. Stepanova, M. Krichevskaya and S. Preis, Catal. Today, 2010, 151, 39–45 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |