
Novel 3-substituted fluorine imidazolium/triazolium salt derivatives: synthesis and antitumor activity†
Abstract
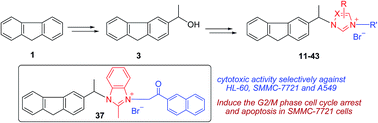
A series of novel (±)-3-substituted fluorene–imidazolium/triazolium salt derivatives has been prepared and evaluated in vitro against a panel of human tumor cell lines. The results suggest that the existence of 2-methyl-benzimidazole or 5,6-dimethyl-benzimidazole rings and substitution of the imidazolyl/triazolyl-3/4-position with a naphthylacyl or 4-methoxyphenacyl group were important for modulating cytotoxic activity. Compounds 37 and 42 were found to be the most potent derivatives with IC50 values of 0.51–2.51 μM and exhibited cytotoxic activities selectively against myeloid leukaemia (HL-60), liver carcinoma (SMMC-7721) and lung carcinoma (A549). Compound 37 can remarkably induce the G2/M phase cell cycle arrest and apoptosis in SMMC-7721 cells. Additionally, compound 30 exhibited selective cytotoxicity to some extent between cancer cells (A549) and normal cells (BEAS-2B).
 Please wait while we load your content...
Please wait while we load your content...

