
On the sintering of gold nanorod assemblies towards continuous networks†
 a
a
Abstract
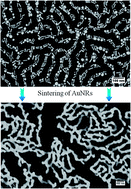
Low temperature sintering of metallic nanoparticles can be used to enhance the thermal properties of composite, thermal interface materials. Here, we present an approach to achieve the coalescence and sintering of a gold nanorod (AuNR) assembly in a block copolymer pattern by thermal annealing at low temperatures. Prior to thermal annealing in a conventional furnace or a rapid thermal processing unit, self-assembly of AuNRs is directed by the guidance of nanochannels in the block copolymer thin films and the removal of surfactants by a brief plasma treatment. Both furnace thermal annealing and rapid thermal annealing are employed to study the sintering behavior of the AuNR assembly. It is found that the sintering process initially takes place locally, resulting in small AuNR aggregates. Eventually the aggregates grow into a globally continuous, percolating network structure. The condensation heat transfer coefficient was measured in an environmental scanning electron microscope by following droplet growth over time and it is shown to be 3.7 times higher for AuNR composites.
 Please wait while we load your content...
Please wait while we load your content...

