
Small lipopeptides possess anti-biofilm capability comparable to daptomycin and vancomycin†
Abstract
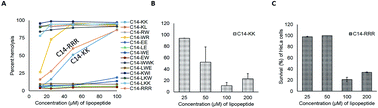
Antibiotic resistance, to a large extent, is related to the formation of bacterial biofilms. Thus, compounds with anti-biofilm capability are of practical importance. Inspired by the recent discovery of two amino acid lipopeptides from marine bacteria, we constructed a family of small lipopeptides with 2–3 amino acids. While no antimicrobial activity was found for anionic lipopeptides, cationic candidates are potent against Staphylococcus strains, such as methicillin-resistant Staphylococcus aureus (MRSA) USA200, USA300, USA400, UAMS-1, Newman, and Mu50. In the simplest design, two lysines (C14-KK) or three arginines (C14-RRR) attached to an acyl chain of 14 carbons were sufficient to make the compounds antimicrobial. These simple lipopeptides are inherently stable towards S. aureus V8 proteinase and fungal proteinase K, more soluble in water, and more selective than other lipopeptides containing a mixture of hydrophobic and cationic amino acids. Furthermore, the activity of C14-RRR was not compromised by salts, serum, or a change in pH. Live cell experiments revealed that these lipopeptides, with a detergent-like structure, killed bacteria rapidly by targeting cell membranes. Importantly, these compounds were also able to inhibit biofilm formation and could even disrupt preformed biofilms of clinically relevant MRSA strains with an in vitro efficacy comparable to daptomycin and vancomycin. These results indicate that small lipopeptides are potentially useful candidates for preventing or eliminating bacterial biofilms alone or in combination with daptomycin or vancomycin.
 Please wait while we load your content...
Please wait while we load your content...

