
Hydrotelluration of acetylenic esters: structural characterization of stereoisomers of methyl/ethyl β-(aryltelluro)acrylates†
Abstract
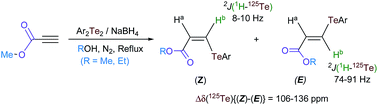
Synthesis and complete characterization of some ester functionalized vinylic tellurides bearing an aryl ligand with varying steric and electronic effects bound to tellurium is described. Hydrotelluration of methyl propiolate using Ar2Te2/NaBH4 in methanol results in a mixture of stereoisomers of methyl β-(aryltelluro)acrylates, ArTeCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOMe (Ar = 4-MeOC6H4, 1A; 1-C10H7, 2A; 2,4,6-Me3C6H2, 3A; C5H5FeC5H4, 4A; 4-Me2NC6H4, 5A; and 2-C4H3S, 6A). The same reaction in ethanol provides isomeric mixtures of the ethyl esters ArTeCHCHCOOEt (1B–6B). However, in the reactions between methyl propiolate and Ar2Te2 (Ar = 2,4,6-Me3C6H2, 4-Me2NC6H4) in isopropanol or t-butanol, no exchange of alkyl groups between the parent ester and the solvent is observed, instead detelluration of the Ar2Te2 to Ar2Te is a competing reaction along with almost exclusive formation of the (Z)-isomers (3Aa, 5Aa). The geometry of the separated stereoisomers is established in solution, with the help of 1H, 13C and 125Te NMR spectrometry. Of particular interest is the observation that 125Te chemical shifts {deshielded in (Z) compared to (E); Δδ = 106–136 ppm} and the geminal heteronuclear coupling constants {2J(1H–125Te) values for (E) are more than seven times that of the corresponding (Z) isomer} can be used to distinguish between liquid isomers. Structural characterization in the solid state by single-crystal X-ray diffraction for the 2Ba, 3Aa, 3Ba, 5Aa, 8 (Z)-isomers as well as for both stereoisomers of 4-Me2NC6H4TeCHCHCOOEt (5Ba and 5Bb) is also presented. The carbonyl O atom of the ester group is invariably involved, at least in the solid state, in a secondary bonding interaction with the Te(II) atom. While an intermolecular Te⋯O interaction gives rise to one-dimensional supramolecular arrays in the crystal lattice of 5Bb with (E) configuration, it is realized intramolecularly in the case of the (Z)-isomers due to the cis position of the chalcogen atoms.
CHCOOMe (Ar = 4-MeOC6H4, 1A; 1-C10H7, 2A; 2,4,6-Me3C6H2, 3A; C5H5FeC5H4, 4A; 4-Me2NC6H4, 5A; and 2-C4H3S, 6A). The same reaction in ethanol provides isomeric mixtures of the ethyl esters ArTeCHCHCOOEt (1B–6B). However, in the reactions between methyl propiolate and Ar2Te2 (Ar = 2,4,6-Me3C6H2, 4-Me2NC6H4) in isopropanol or t-butanol, no exchange of alkyl groups between the parent ester and the solvent is observed, instead detelluration of the Ar2Te2 to Ar2Te is a competing reaction along with almost exclusive formation of the (Z)-isomers (3Aa, 5Aa). The geometry of the separated stereoisomers is established in solution, with the help of 1H, 13C and 125Te NMR spectrometry. Of particular interest is the observation that 125Te chemical shifts {deshielded in (Z) compared to (E); Δδ = 106–136 ppm} and the geminal heteronuclear coupling constants {2J(1H–125Te) values for (E) are more than seven times that of the corresponding (Z) isomer} can be used to distinguish between liquid isomers. Structural characterization in the solid state by single-crystal X-ray diffraction for the 2Ba, 3Aa, 3Ba, 5Aa, 8 (Z)-isomers as well as for both stereoisomers of 4-Me2NC6H4TeCHCHCOOEt (5Ba and 5Bb) is also presented. The carbonyl O atom of the ester group is invariably involved, at least in the solid state, in a secondary bonding interaction with the Te(II) atom. While an intermolecular Te⋯O interaction gives rise to one-dimensional supramolecular arrays in the crystal lattice of 5Bb with (E) configuration, it is realized intramolecularly in the case of the (Z)-isomers due to the cis position of the chalcogen atoms.
 Please wait while we load your content...
Please wait while we load your content...

