
Influence of the carbon nanotube surface modification on the microstructure of thermoplastic binders
S. V. Larina,
A. D. Glovab,
E. B. Serebryakovb,
V. M. Nazarycheva,
J. M. Kennyacd and
S. V. Lyulin*ab
aInstitute of Macromolecular Compounds, Russian Academy of Sciences, Bol'shoi pr. 31 (V.O.), St. Petersburg, 199004, Russia. E-mail: s.v.lyulin@gmail.com; Fax: +7 812 328686; Tel: +7 812 3285601
bDepartment of Physics, St. Petersburg State University, Ul'yanovskaya str. 1, Petrodvorets, St. Petersburg, 198504, Russia
cMaterials Science and Technology Centre, University of Perugia, Loc. Pentima, 4, Terni, 05100, Italy
dInstitute of Polymer Science and Technology, ICTP-CSIC, Juan de la Cierva, 3, Madrid, 28006, Spain
First published on 3rd June 2015
Abstract
The structural properties of polymer nanocomposites based on thermoplastic polyimides filled with surface-modified carbon nanotubes (CNT) have been studied by means of fully-atomistic molecular-dynamics simulations. The influence of the distribution of functional carboxyl groups over the CNT surface on the polymer-matrix density distribution, and the orientational ordering of polymer chains have been investigated. It was shown that the polymer shifts far away from the nanoparticle surface with increase of the CNT modification degree. The orientational ordering of PI chains was not observed in the case of nanocomposites filled with modified CNTs where carboxyl groups are distributed uniformly on the surface. However, in case of the edge-modified CNTs the polymer can interact with the CNT surface; such edge-modified nanoparticles induce orientational ordering of crystallisable polyimide chains which can be considered as an initial stage of the polymer matrix crystallization.
Introduction
Reinforcement of a polymer matrix with nanofillers is regarded as one of the most promising ways to develop new high-performance materials. This is due to the fact that even in the case of a small volume fraction of the nanofiller, the interface area between the composite components can be large enough to cause the required changes in the polymer binder properties.The use of functional polymers with a rather complicated structure combining high thermal properties (high glass transition and thermal destruction temperatures) with advanced mechanical, electrical and optical properties as polymer binders, is a very promising method to create novel composite materials.1–5 Among such polymers, thermoplastic aromatic polyetherimides play a key role.6–9 Their use allows the utilization of ecologically safe melting technologies in order to create composites and nanocomposites. This simplifies significantly the processing compared to the loading of nanofiller particles into the polymer binder during polymerization.10–13
Subject to the chemical composition of polyimides (PI), their structural and physical properties may vary a lot. In particular, some polyimides may crystallize.14–19 The crystallization of polyimides is relevant in terms of technology, since the increase in the crystallinity will lead to a general enhancement of the mechanical performance of the crystallized polyimides, despite possible side effects impairing the properties. Therefore, it is of importance to understand the conditions of formation of crystalline domains in polyimides and polyimide-based composites depending on the polymer chemical structure and type of nanoparticles used as fillers.
Carbon nanotubes (CNT) represent one of the most wide-spread nanofiller together with graphene and fullerenes.4 It is well-known that the loading of carbon nanofillers in thermoplastic polyimides often leads to enhancing their mechanical and thermal properties.20–22 However, the mechanism of these changes is not completely clear since CNT loading into the polyimide matrix as such does not necessarily mean significant improvement of the composite properties, compared to those of the unfilled polymer.23 Furthermore, nanotubes may display a rather small affinity to polyimides. In this case the mechanism of improvement of the composite mechanical performance due to the load transfer to the filler may be ineffective. Yet it is well-known that both graphene and CNT can initiate the crystallization of various polymers, including polyethylene,24–27 polycaprolactone,28 polypropylene,25,29–31 polyvinyl alcohol,32 polylactic acid,33,34 polyamides,25 polyetheretherketone,35,36 polyacrylonitrile,37 poly(vinilidene fluoride)38 as well as heterocyclic polyalkylthiophenes39 and aromatic polyimides.14–19 It is interesting that nanotubes can even lead to the crystallization of such polyimides as ODPA-P3 which normally do not crystallize in bulk.14
Therefore, the addition of a CNT to the crystallisable thermoplastic PI causes noticeable changes in the polymer binder properties related to the changes in its structure,14–19 which modify the mechanical and thermal behaviour of the composite, and should be taken into account when developing new polymer materials.25,40
The efficiency of the CNT loading into the polymer may be reduced a lot as a result of their tendency to aggregation.38,41,42 To prevent CNT aggregation, their surface is modified with functional groups increasing the affinity of the nanofiller and the polymer matrix.38,43–45 The CNT surface modification usually starts with attaching hydroxyl (–OH) or carboxyl (–COOH) groups to it. As a rule, they attach themselves to the nanotube surface during the oxidation with oxygen, air, aqueous solution of hydrogen peroxide, concentrated sulfuric acid, nitric acid or mix of acids.46 The number of –COOH and –OH groups on the CNT surface increases with growing temperature during the acid treatment as well as in the case of the extended treatment duration.38,47,48 The number of functional groups attached to the surface also depends on the oxidation procedure and oxidizing agents.
Preferable sites for the hydroxyl and carboxyl groups to attach to the CNT are carbon atoms at the CNT edges48 and CNT surface defects which represent inclusions of five or seven-membered carbon rings into the hexagonal grid made of carbon atoms constituting the CNT surface, and other parts where CNT carbon atoms change sp2-hybridization to sp3-hybridization. Such defects can be created intentionally during the distorting functionalization of the CNT surface. In this case, defective sites of the surface usually contain about 5–10% of the CNT atoms.49 The distorting modification does not change electrical and mechanical properties of the tubes,46 which is why the CNT defects play an important part since they can be used as a basis for the further CNT surface functionalization.
Attachment of carboxyl groups to the CNT surface offers a number of advantages compared to that of other groups, because they can be used for further nanotube functionalization. Presence of –COOH groups on the CNT surface makes it possible to attach organic or inorganic molecules,47,48,50,51 including the binder oligomers, which is important for changing the CNT solubility in the solutions of low-molecular and high-molecular compounds and for improving the dispersion in nanocomposites. Therefore, the functionalization of the carbon nanotube surface with –COOH groups is an important stage to prevent their aggregation in the polymer matrix.
Since it is assumed that the nucleation ability of carbon fillers are related to the interaction of the polymer matrix chains with the filler surface which are determined for heterocyclic polymers by π–π interactions, whereas in the case of the above mentioned surface modification, it is possible that the crystallization is not initiated, which can be critical for maintaining the required composite properties. Indeed, the study by Liang et al.33 carried out for polylactic acid-based composites shows that the functionalization of the nanotube surface leads to the loss of CNT nucleation activity. The changes observed are attributed to the decrease in the filler surface area available for the contact with polymer chains, which is related to the steric effect of functional groups coupled with the nanotube.
Molecular-dynamics (MD) atomistic simulations are the most efficient methods to study the phenomena at the atomistic level which take place during the nanoparticles loading into the polymer matrix, and to establish the mechanism of the associated changes. Such simulation enables us to explore structural properties of the systems under scrutiny on the atomistic scale, and establish the mechanisms stipulating the changes in the polymer properties after nanofiller loading.51–56
Composites based on thermoplastic PIs R-BAPB and R-BAPS filled with carbon nanotubes52 or graphene53 have been studied in our previous papers using atomistic computer simulations on a microsecond time scale. It was shown that the R-BAPB-based composites featured the formation of ordered structures, both near the filler surface and at a some distance. The ordering effect can be interpreted as the initial stage of the polymer crystallization induced by the nanofiller.57 This process is caused by the ordered alignment of subsequent flat phenylphthalimide (PPI) and diphenyl (DP) fragments of the R-BAPB polyimide repeating unit (Fig. 1),52,53 resulting from the interaction with the filler surface.
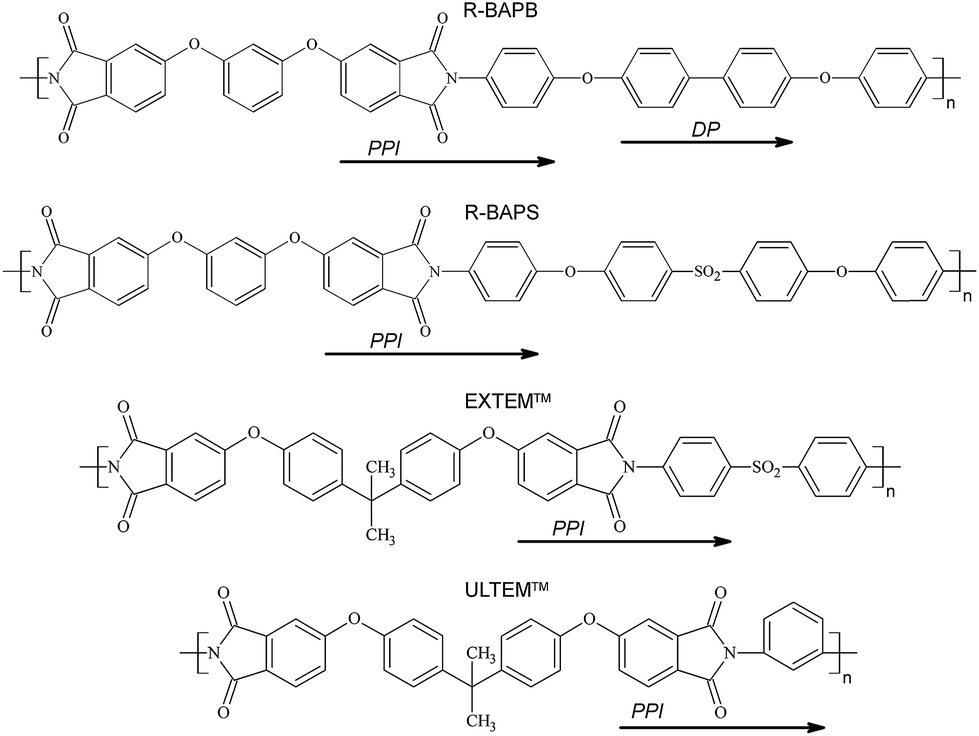 | ||
| Fig. 1 Chemical structures of polyimides R-BAPB, R-BAPS, EXTEM and ULTEM (top down). Arrows indicate flat phenylphthalimide (PPI) and diphenyl (DP) fragments of the polyimide monomer units. | ||
This study aims to investigate the influence of the most wide-spread type of the CNT surface modification with carboxyl groups on a CNT ability to act as a crystallization nucleant. This possibility is related to both the position of the modifying carboxyl groups on the CNT surface, and to the CNT surface area remaining available for the interaction with the polymer chains. It can be assumed that in the case of a certain grafting density of functional groups onto the CNT surface or in the case of their certain distribution on the surface, the influence of steric limits on the polymer interaction with the CNT will be reduced, and the initiation of crystallization with modified nanotubes can be similar to the case of a non-modified filler loading. These effects will be studied in the present paper in the nanocomposites based on thermoplastic polyimides filled with CNTs with a surface variously modified by carboxyl groups. First, the CNTs modified with carboxyl groups uniformly distributed on the nanotube surface were taken into account. However, as the preferential direction of carboxylic groups attachment to CNT is terminal atoms,48 the more realistic model with carboxyl groups attached to the CNT ends was investigated also.
The simulation of the systems based on several various polymer binders enables us to determine the influence of the polymer chemical composition on their structural behaviour in the nanocomposites. Obtained results may help to establish the interrelation between composite properties and the chemical structure of its components, which is of relevance for the developing of new types of composite materials and polymer binders for them.
The second section of this paper deals with the model and simulation approaches used in the study. In the third section we discuss the influence of the modified carbon nanotubes loading on the structural properties of polyimides near the filler surface, subject to the polyimide chemical composition, modification degree of the carbon nanotube and the distribution type of functional groups on the CNT surface. Particular emphasis is laid on the ability of modified CNTs to initiate the ordering of polyimide chains near the nanoparticle surface. The final section contains major conclusions made on the basis of the obtained results.
Model and simulation method
The study addresses nanocomposites based on heat-resistant thermoplastic polymers. Two main types of polyimides for matrices are considered which are synthesized and actively explored in the Institute of Macromolecular Compounds of the Russian Academy of Sciences:11,13,15–19,58,59 crystallisable R-BAPB based on 1,3-bis-(3′,4-dicarboxyphenoxy)-benzene (dianhydride R) and 4,4′-bis-(4′′-aminophenoxy)-diphenyl (diamine BAPB) and amorphous R-BAPS based on dianhidride R and 4,4′-bis-(4′′-aminophenoxy)-diphenylsulfone (diamine BAPS), whose chemical structures are given in Fig. 1. These polyimides have identical dianhydride fragments in repeating units and differ in the diamine fragment composition. The R-BAPS diamine has an additional hinge group SO2 between benzene rings. Such modification of the chemical structure enhances the flexibility of polymer chains of R-BAPS compared to that of R-BAPB; at the same time it leads to larger glass transition temperature of R-BAPS60–62 due to the increased contribution of dipole–dipole interactions into the total system energy, related to the presence of the polar sulfone group in the R-BAPS repeating unit, as was demonstrated in our previous papers.58,63–65 The increased flexibility of the chain, and enhanced dipole–dipole interactions in the PI R-BAPS, as opposed to the R-BAPB, prevent crystallization of this polymer even in the presence of carbon nanoparticles which can act as good nucleants. Notably, the loading of modified CNTs with polarized carboxyl groups on the surface into the R-BAPS may have a different influence on the polymer structure, compared to that of the R-BAPB polyimide where electrostatic interactions do not play such a significant role as in the case of the R-BAPS. Therefore, the comparison of the structure of composites based on the polyimides containing polar groups in the repeating unit with composites based on PIs without polar groups allows to evaluate the significance of electrostatic interactions for the ordering processes occurring in the polymer matrix after the nanofiller loading.For reference, we additionally simulated composites which are based on commercial heat-resistant polyimides ULTEM and EXTEM produced by SABIC Innovative Plastics.66–70 Similar to the case of R-BAPB and R-BAPS polyimides, the major difference in their chemical structure (shown in Fig. 1 as well) is the diamine modification with the presence of a sulfone group in the EXTEM polyimide. Thus, in the case of EXTEM, dipole–dipole interactions between the polar groups of polymer chains and of the CNT surface may impact the polymer matrix structure in the nanocomposites under the study. Generally, comparison of the simulation results for polyimides R-BAPB and R-BAPS with the results obtained for ULTEM and EXTEM will enable us to establish the influence of electrostatic interaction on the structural performance of nanocomposites with modified CNTs.
Simulation of the composites based on crystallisable polyimide R-BAPB with CNT which differ in the degree of the surface modification by functional groups ωM and their location make it possible to determine the influence of the CNT surface modification on the orientation ordering processes of the polymer chains near the filler surface which can be regarded as the initial stage of the polymer crystallization.52,53
Similar to our previous paper52 carbon nanotubes of 4.7 nm long with chirality (5, 5) were used as fillers with carboxyl groups attached to the CNTs. To determine the influence of the degree of the CNT surface modification on the microstructure of the polymer matrix in the nanocomposite, nanotubes were explored with various number of attached carboxyl groups corresponding to different surface modification degrees ωM = 5% and 10%. In the first case, 20 carboxyl groups were attached to the CNT surface that have 400 carbon atoms, and 40 carboxyl groups were attached in the last one (Fig. 2). The positions of the groups were chosen so that they were distributed more or less uniformly on the carbon nanotube surface, that is, with similar intervals between them both along the nanotube axis and in the tangential direction. It was found that the simplest location variant with regard to these requirements was the position of carboxyl groups in rings equally spaced along the CNT axis, with each ring containing 5 carboxyl groups, which may be considered as a first approximation to the uniform distribution.
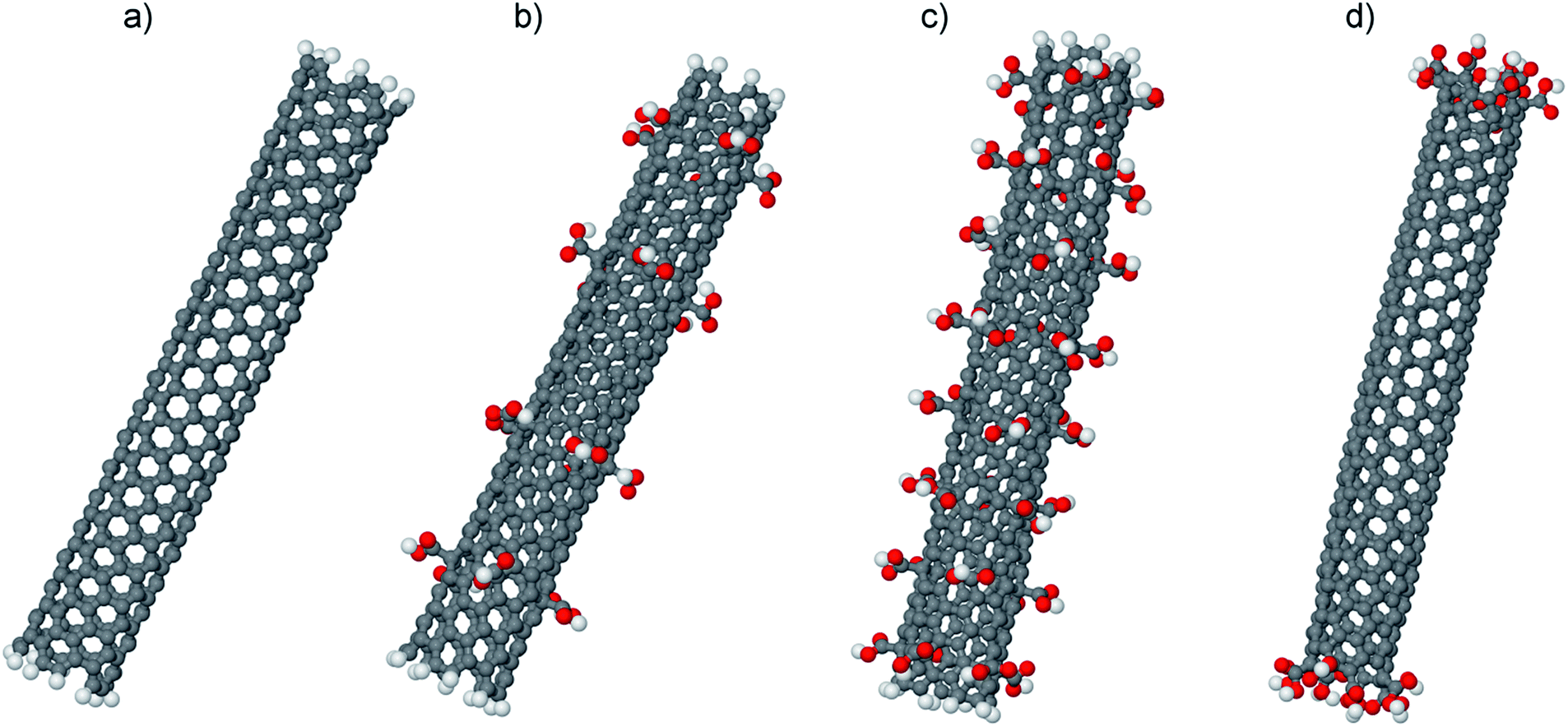 | ||
| Fig. 2 CNT configurations with (a) non-modified surface and with different degree of modification with carboxylic groups ωM = 5% (b) and 10% (c); (d) configuration of CNT with carboxylic groups attached to the edge carbon atoms (edge-modified CNT). | ||
The fully-atomistic model and simulation approach used for this research were proved in our previous studies exploring the structural and thermal properties of the heat-resistant polyimides58,60–65 and composites based on them.52,53
The computer simulation of the systems under the study was performed with the Gromacs computational package71 using the Gromos53a5 force field.72 The Gromacs package allows for computer simulation using atomistic models on a microsecond time scale, which is required for the investigation of initial stages of the structural ordering of crystallisable polyimides near the carbon nanoparticle surface.
Similar to our previous studies52,58,60–65 the systems considered contain one CNT and 27 polyimide chains with a polymerization degree of n = 8 (for R-BAPB and R-BAPS) or n = 9 (for ULTEM and EXTEM), which corresponds to the beginning of so-called “polymer regime” in thermal properties with molecular mass around 6–6.5 kg mol−1.52,59,60,64
The simulation was performed in the NpT ensemble at a temperature of 600 K under a pressure of 1 bar. The pressure and temperature were maintained at the required level using the Berendsen thermostat and barostat;73 the time constants for which were taken as τT = 0.1 ps for the thermostat, and τP = 0.5 ps for the barostat. Test simulations showed that the use of the Nose–Hoover thermostat and the Parhinello–Rahman barostat do not lead to any significant changes in the discussed structural properties. To maintain the set lengths of the bonds between the atoms, the LINCS algorithm was used.74
The values of the partial charges for the atoms of carboxyl groups attached to the CNT were taken as standard values for the Gromos53a5 force field72 which was parameterized for simulating systems in the media with low permittivity. Parametrization of the charges in this force field was done using the HF/6-31G* method.72,75 The same method was used to calculate the partial atomic charges in the considered polyimides. The charge distribution was performed using the Mulliken method. Such parametrization of electrostatic interactions enables us to reproduce experimental values of the thermal properties of the heat-resistant PIs with a high degree of accuracy for the set of PIs.58,62,65 The calculated partial charges were published in our previous studies.52,64 Ewald summation method (PME)76 was used to account for the electrostatic interactions.
As was mentioned above, the simulation was performed within the microsecond time scale similar to our previous studies.52,53,60–65 The equilibration procedure included stages of the system compression from the initial low-density configuration (polymer gas), cyclic annealing within the temperature range of 600 to 290 K, equilibration at a high temperature for 1.5 μs, and further production run during 1 μs. The given stages were carried out without accounting for electrostatic interactions (EI). In this case, the simulation times of about 1 μs are close to the typical time of the polymer chain centre of mass displacement to the distance comparable to the chain size, which enables us to obtain the well-equilibrated configurations.52,62
Since the presence of EI slows down the diffusion of the polyimide chains in the melt approximately by two orders of magnitude,52,61,62 the simulation with switched-on electrostatics was performed using the equilibrated configurations of the systems with switched-off electrostatics. To this end, the system configuration was chosen which were obtained after the described above simulation for 2.5 μs without electrostatics. Then these systems were simulated with switched-on partial charges for at least 100 ns. The specified period of the simulation with electrostatic interactions is characterized by the relaxation of density and local structure of the systems under the study. The observable changes in nanocomposite structure, i.e. the change of density distribution, occur during first 10 ns of simulation after switching on electrostatic interactions. At the same time the chain size and shape do not change sufficiently during simulation with partial charges switched-on. Thus, we suppose that 100 ns simulation with accounting for EI after long-time equilibration without EI is enough to obtain reliable data on structural properties of the nanocomposites studied. The previous research demonstrated that the account of electrostatic interactions has a rather little influence on the global structural order on the composites under scrutiny.52 Final configurations were subsequently used for analysis.
To characterize structural properties of composites based on modified nanotubes we calculated density distributions of polyimides as a function of distance from the nanotube axis ρ(r) and pair polyimide-nanotube distribution functions gPI-CNT(r) similar to the composites with pristine CNTs.52 We also calculated the orientation parameters of flat PPI fragments of the polyimide monomer units (Fig. 1): distribution of the angle θ between the PPI fragment of the monomer unit and the nanotube axis subject to the distance between them, and the relevant order parameter S(r):
S(r) = 3/2〈cos2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ(r)〉−1/2, θ(r)〉−1/2,
| (1) |
θ(r)〉 is the mean square cosine of the angle between the nanotube axis and the polyimide monomer unit fragment located at a distance r from the nanotube axis. The angle θ is determined as shown in Fig. 3.
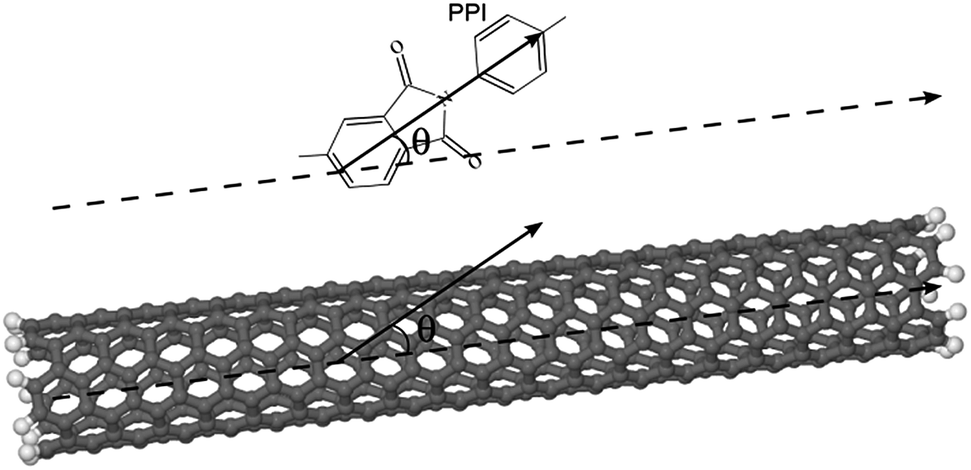 | ||
| Fig. 3 Determination of the angle θ between the flat PPI fragment of the polyimide repeat unit and the carbon nanotube axis. | ||
The polyimide density distribution relative to nanotube axis ρ(r) is calculated as following
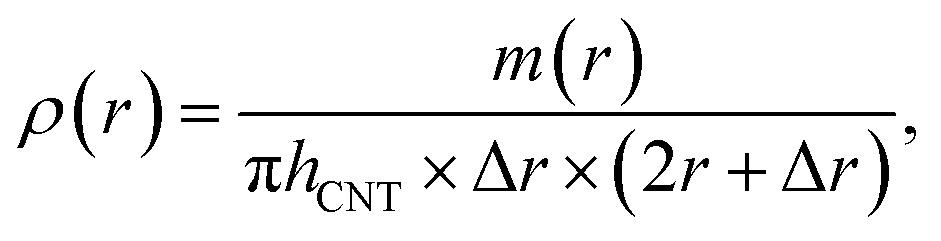 | (2) |
The pair correlation functions gAB(r) describes the local distribution density of the component B near the component A in relation to the mean density of the component B in the system:
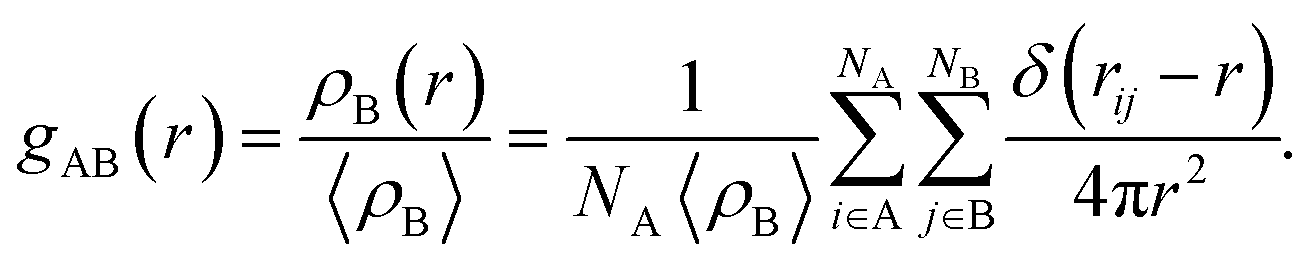 | (3) |
Resulted pair correlation functions make help to understand the distribution of various components in the composite. Such analysis is widely used for the systems in a condensed state, including composite materials.54,55,77–80
The simulations performed with various starting points give similar results on the properties investigated. We estimate run-to-run variations of the results to be less than 5%.
Results
Density distribution and pair distribution functions
The PIs density distribution, eqn (2), in nanocomposites with CNTs with various degrees of the surface modification by carboxyl groups is shown in Fig. 4. In the case of composites with pristine nanotubes (ωM = 0%) a dense polymer layer appears around the filler at a distance of r ∼ 0.7 nm away from the CNT axis. For composites containing a CNT with ωM = 10%, this subsurface polymer layer shifts to a distance of r ∼ 0.9 nm. This shift is related to the impossibility for the polyimide to reach the nanotube surface due to the steric limits, related to the presence of high-volume carboxyl groups on the CNT surface. Meanwhile, in the case of composites containing CNTs with intermediate value of ωM = 5%, the ρ(r) dependence demonstrates that the polyimide chain fragments may be located either at the same distance as in the case of pristine CNTs, or far from it corresponding to the fist peak of ρ(r) in case of ωM = 10%. Consequently, in the case of a CNT with ωM = 5%, the polymer density distribution displays a typical bimodal behavior. These curves are characterized, first of all, by the maximum at r ∼ 0.7 nm, and, secondly, a shoulder or second maximum at r ∼ 0.9 nm. The shoulder corresponds to the shifting of the polyimide far away from the CNT surface, which is facilitated by carboxyl groups on the surface.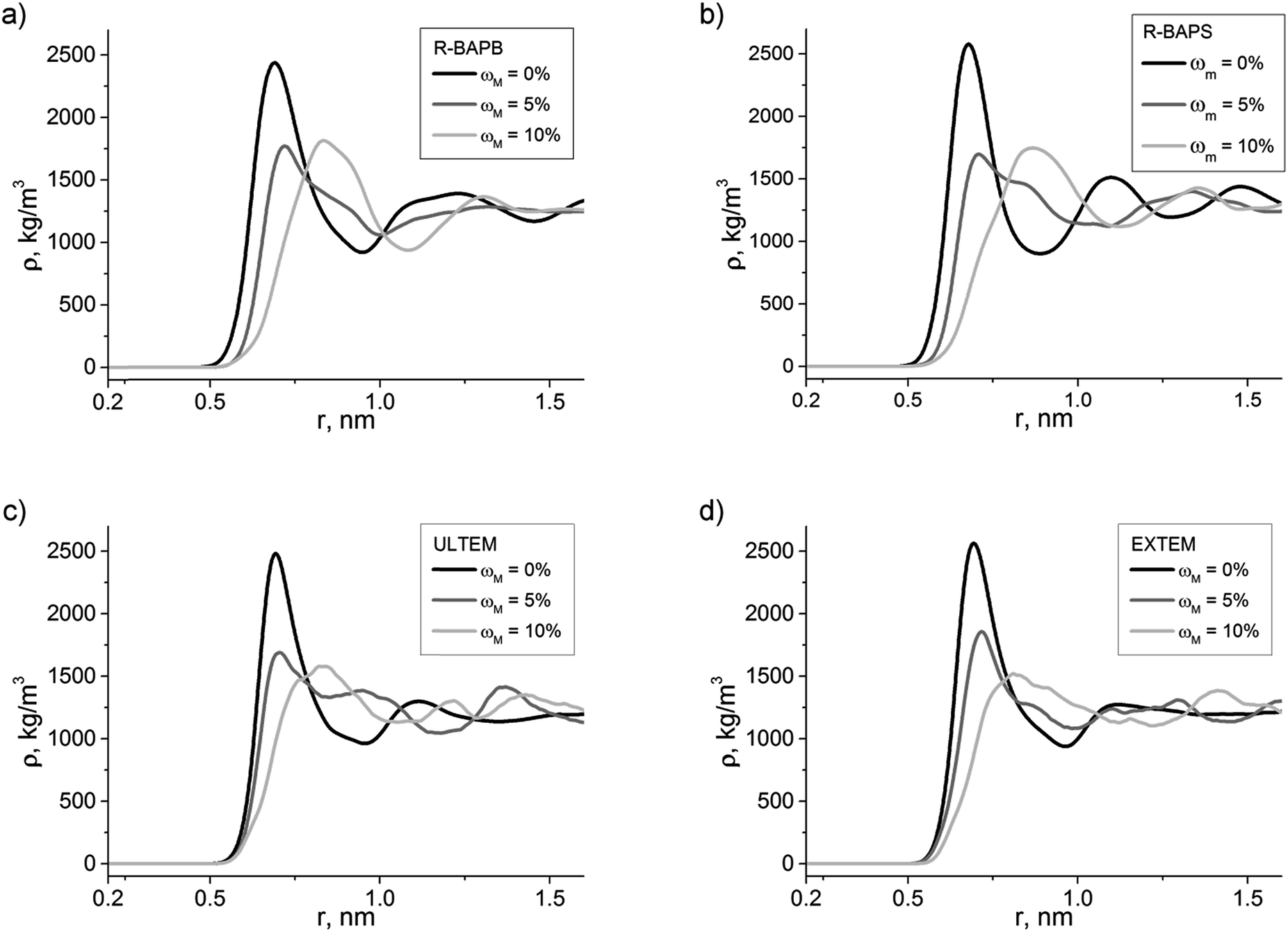 | ||
| Fig. 4 Density distribution for polyimides (a) R-BAPB, (b) R-BAPS, (c) ULTEM, and (d) EXTEM in the composites based on them, filled with nanotubes with various surface modification degrees (with switched-on electrostatics). ωM = 0% corresponds to pristine CNT. | ||
Notably, the general patterns of the curves describing the density distribution of various polyimides in nanocomposites with modified CNTs do not differ from each other, similar to the case of composites containing pristine CNTs.52 This is due to the fact that the atomic composition of the polymers under study is almost identical. The only significant distinction of the density distribution is that in the case of polyimides containing polar sulfone groups (R-BAPS and EXTEM), the density distribution maxima are narrower and are located at a smaller distance from each other, which is associated with additional compaction of these polymers due to the dipole–dipole interactions.
The pair distribution functions, see eqn (3), for the PI atoms relative to the carbon nanotube atoms gPI-CNT(r) were calculated for all considered nanocomposites. All the polyimides feature the identical general pattern of the pair distribution functions and the nature of the changes in composites with various CNTs (see Fig. 5, where the pair distribution functions gPI-CNT(r) for nanocomposites based on the polyimide R-BAPB are shown as an example). The increase of the CNT surface modification degree leads to the disappearance of the first maximum or shoulder in the pair distribution function gPI-CNT(r) corresponding to the subsurface polyimide layer, and to the general shift of gPI-CNT(r) towards larger values of distance r due to the growing effective radius of the CNT with a modified surface. These results confirm the previous conclusion that the increasing of modification degree of the nanotube surface leads to the polymer shifting far away from the CNT axes.
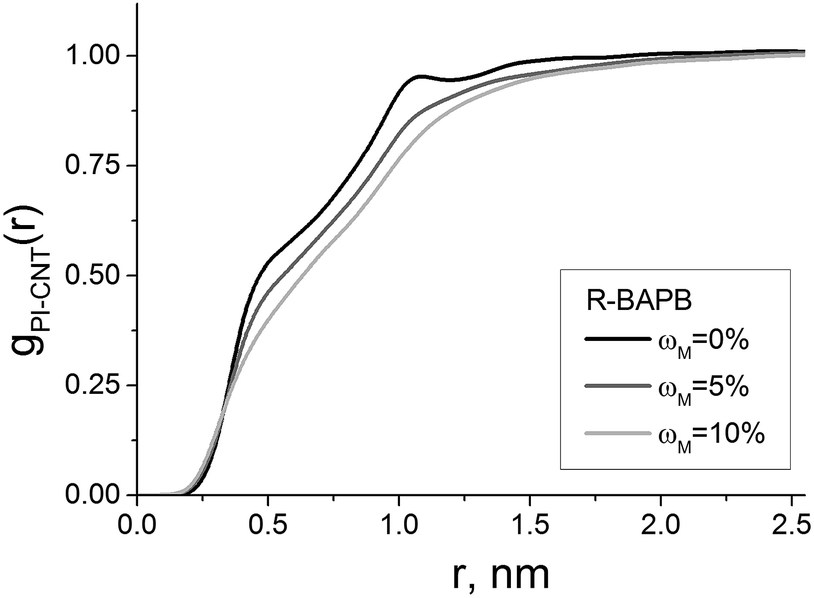 | ||
| Fig. 5 Pair distribution functions gPI-CNT(r) of the R-BAPB polyimide atoms in relation to the CNT atoms in nanocomposites with switched-on electrostatics filled with nanotubes with various surface modification degrees ωM. | ||
Orientation of flat fragments
Similar to the case of nanocomposites filled with pristine CNTs,52 the calculation of the orientation characteristics (order parameters and distribution of the orientation angles relative to the nanotube axis) of the various PI chain fragments for systems with switched-on EI showed that they are qualitatively the same as the results obtained without electrostatics. Also the values of the orientation characteristics for these systems display rather strong fluctuations52 due to the mobility reduction of polymer chains observed in the simulation with EI switched-on.52,61,62 Therefore, all orientation characteristics of polyimide chains shown further in this section have been obtained for the systems simulated without electrostatic interactions, for the clarity.The analysis of the order parameters S(r) describing the orientation of the PPI flat fragments in nanocomposites with various CNT surface modification degrees (Fig. 6) demonstrates that the increased ωM results in the reduction of the ordering degree of the PPI fragments relative to the nanotube axis. For polyimides R-BAPB and R-BAPS, the maxima tend to disappear on the curve S(r) at the any ωM, which is related to the lack of PI chains ordering along the nanotube axis even in immediate proximity to the CNT surface (Fig. 7). Fig. 7 also shows that composites containing CNTs with ωM = 10% have no dominating orientation direction, even for the chain fragments located closest to the nanotube axis. Similar results were obtained for composites based on all the polyimides under study as well. Notable, polyimides ULTEM and EXTEM do not show any significant ordering of the chain flat fragments near the nanotube even in the case of composites with a pristine CNT.
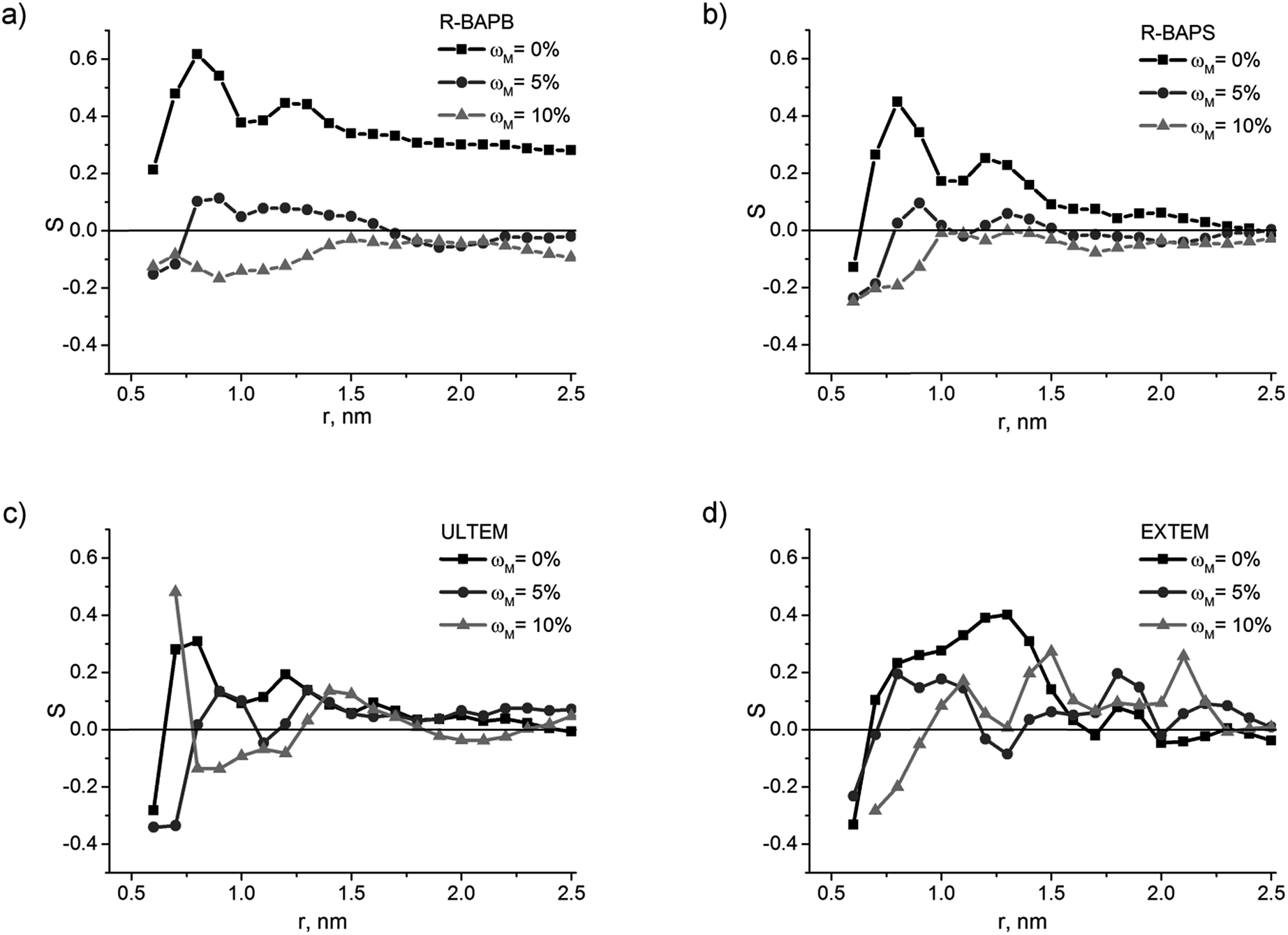 | ||
| Fig. 6 The order parameter S(r) of the PPI flat fragment in composites based on polyimide (a) R-BAPB, (b) R-BAPS, (c) ULTEM, and (d) EXTEM filled with CNTs with different surface modification degrees ωM. | ||
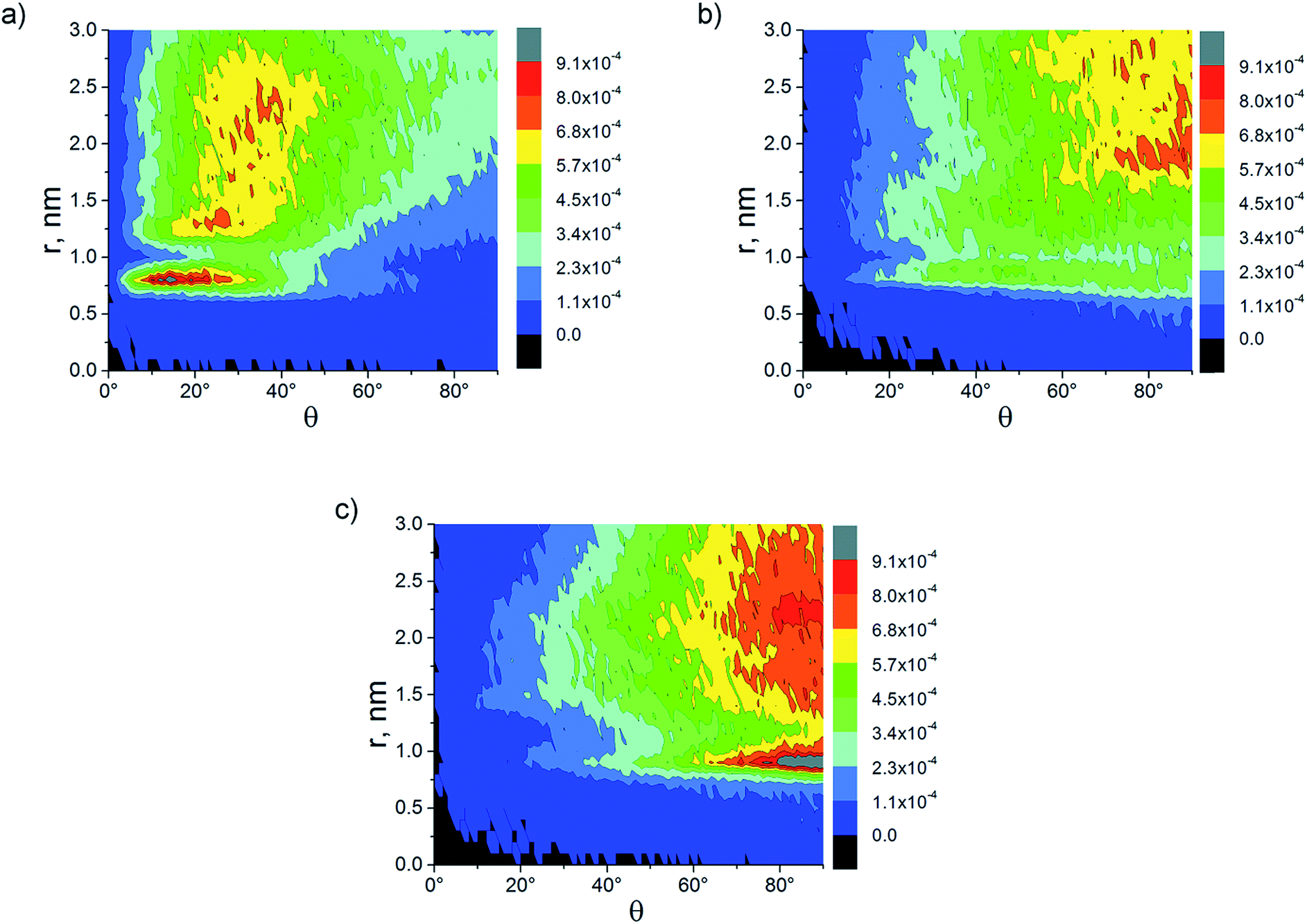 | ||
| Fig. 7 Distributions of orientation angles of the PPI flat fragment in nanocomposites based on crystallisable polyimide R-BAPB filled with a CNT with a different surface modification degree (a) ωM = 0%, (b) ωM = 5%, and (c) ωM = 10%, obtained after the simulation for 2.5 μs without electrostatic interactions. | ||
Therefore, we can conclude that the modification of the CNT surface with carboxyl groups leads to the decrease in the nucleation ability of carbon nanofiller. This may be attributed to the fact that the carboxyl groups located on the CNT surface prevent the PI chains from interacting with unmodified sites of the filler surface, which reduces the CNT nucleating effect.
A possible solution to avoid this modified CNT effect is increasing the unmodified CNT surface area available for the interaction with PI chains. In the model used, it corresponds to the increase of distance along the nanotube axis between the rings of modifying groups. To verify this assumption we have simulated additionally nanocomposites based on the crystallisable polyimide R-BAPB containing modified CNTs with functional groups attached only to the carbon atoms at the CNT edges (Fig. 2d). The surface modification degree of such a CNT equals 5% as well, yet the surface area available for the interaction with PI chains is substantially higher than that of modified CNTs with functional groups uniformly distributed over the surface. Structural properties of such composites are shown in Fig. 8.
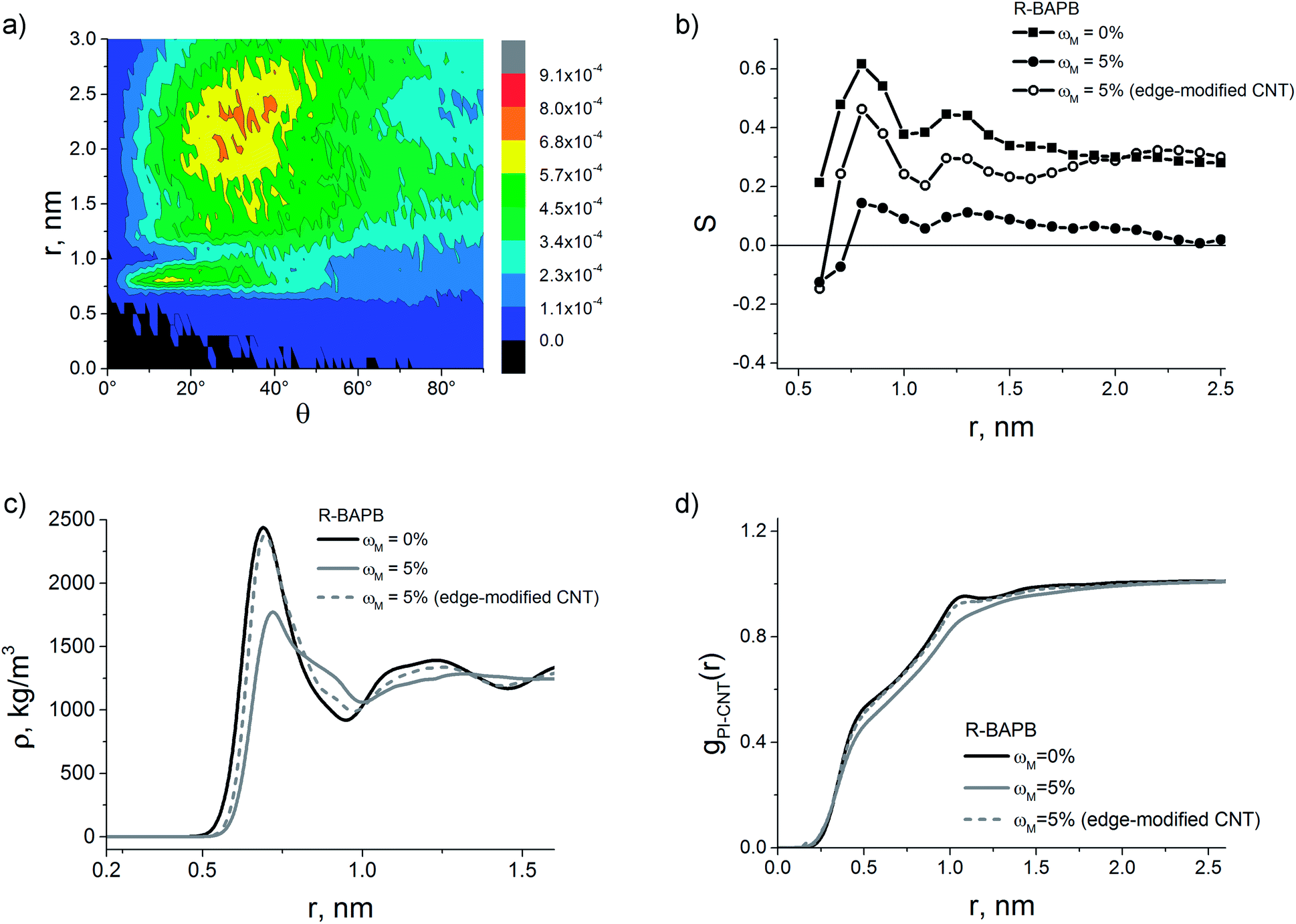 | ||
| Fig. 8 (a) Orientation angle distribution of the PPI flat fragment in the composites based on the R-BAPB polyimide filled with CNTs modified with carboxyl groups at the terminal carbon atoms, with a surface modification degree of 5%, and (b) order parameters for R-BAPB based nanocomposites. (c) Polyimide density distribution in composites relative to the CNT axis. (d) PI-CNT pair distribution functions. | ||
In the case of carboxyl groups attached to the terminal carbon atoms of CNT, the PI density distributions practically coincide in composites with modified and with pristine CNTs. Furthermore, the edge-modified CNT loading into the matrix does not significantly change the pair distribution functions, testifying the negligible influence of such modification on the polymer interaction with the nanotube surface and suggesting the possible formation of ordered structures in composites based on the R-BAPB polyimide filled with such CNTs.
Indeed, the analysis of the orientation angle distributions of the PPI flat fragments of the R-BAPB polyimide relative to the nanotube axis and the corresponding order parameter, showed that composites filled with edge-modified CNTs display the formation of a structure with the PI chain flat fragments oriented along the CNT, both near the filler surface and at a distance from it, similar to composites with pristine CNTs.52
Conclusions
We have simulated the structural properties of various heat-resistant polyimides in composites filled with carbon nanotubes with a surface modified by carboxyl groups. The dependence of the structural organization of polyimide chains on the CNT surface modification degree and the location of the functional groups over the surface have been studied.It is shown that the CNT surface area available for contacts with PI matrix influences significantly the polymer matrix structure. Increase of CNT surface modification degree leads to reduced surface area of the filler available for the interaction with polymer chains. Thus, in the case of uniform distribution of functional groups on the CNT surface no orientation ordering of the flat fragments of polyimide chains have observed even near the filler surface.
On the other hand, in the case of edge-modified CNT the most of the CNT surface can interact with crystallisable R-BAPB matrix providing its ordering and further crystallization.
Acknowledgements
The study was carried out with the financial support from the Ministry of Education and Science of the Russian Federation under the Contract no. 14.Z50.31.0002 (megagrant of the Government of the Russian Federation according to the Resolution no. 220 of April 9, 2010).Notes and references
- A. Baker, S. Dutton and D. Kelly, Composite materials for aircraft structures, American Institute of Aeronautics and Astronautics Inc., Reston, Virginia, 2nd edn, 2004 Search PubMed.
- A. P. Mouritz and A. G. Gibson, Fire properties of polymer composite materials, Springer, Dordrecht, 2006 Search PubMed.
- D. Gay, S. V. Hoa and S. W. Tsai, Composite materials. Design and application, CRC Press, London, 2003 Search PubMed.
- L. Peponi, D. Puglia, L. Torre, L. Valentini and J. M. Kenny, Mater. Sci. Eng., R, 2014, 85, 1 CrossRef PubMed.
- G. O'Bryan, B. M. Wong and J. R. McElhanon, ACS Appl. Mater. Interfaces, 2010, 2, 1594 Search PubMed.
- M. I. Bessonov, M. M. Kotton, V. V. Kudryavtsev and L. A. Lajus, Polyimides – thermally stable polymers, Consultants Bureau, New York, 1987 Search PubMed.
- Polyimides: Fundamentals and Applications, ed. M. K. Ghosh and K. L. Mittal, Marcel Dekker Inc., New York, 1996 Search PubMed.
- H. Ohya, V. V. Kudryavtsev and S. I. Semenova, Polyimide Membranes-Applications, Fabrication and Properties, copublished by Kodansha Ltd., Gordon and Breach Science Publishers, S.A., Tokyo and Amsterdam, 1996 Search PubMed.
- M. J. M. Abadie and A. L. Rusanov, Practical Guide to Polyimides, Smithers Rapra Technology Limited, Shawbury, 2007 Search PubMed.
- Multifunctional Polymer Nanocomposites, ed. J. Leng and L. A. Kin-tak, CRC Press, London, 2010 Search PubMed.
- V. E. Yudin and V. M. Svetlichnyi, Russ. J. Gen. Chem., 2010, 80, 2157 CrossRef CAS.
- H. Xu, H. Yang, L. Tao, J. Liu, L. Fan and S. Yang, High Perform. Polym., 2010, 22, 581 CrossRef CAS PubMed.
- V. E. Yudin, G. M. Divoux, J. U. Otaigbe and V. M. Svetlichnyi, Polymer, 2005, 46, 10866 CrossRef CAS PubMed.
- M. Hegde, U. Lafont, B. Norder, S. J. Picjen, E. T. Samulski, M. Rubinstein and T. Dingemans, Macromolecules, 2013, 46, 1492 CrossRef CAS.
- V. E. Yudin, V. M. Svetlichnyj, A. N. Shumakov, R. Schechter, H. Harel and G. Marom, Composites, Part A, 2008, 39, 85 CrossRef PubMed.
- T. Kurose, V. E. Yudin, J. U. Otaigbe and V. M. Svetlichnyj, Polymer, 2007, 48, 7130 CrossRef CAS PubMed.
- V. E. Yudin, V. M. Svetlichnyi, A. N. Shumakov, D. G. Letenko, A. Y. Feldman and G. Marom, Macromol. Rapid Commun., 2005, 26, 885 CrossRef CAS PubMed.
- V. E. Yudin, A. Y. Feldman, V. M. Svetlichnui, A. N. Shumakov and G. Marom, Compos. Sci. Technol., 2007, 67, 789 CrossRef CAS PubMed.
- V. E. Yudin, V. M. Svetlichnyi, G. N. Gubanova, A. L. Didenko, T. E. Sukhanova, V. V. Kuidryavtsev, S. Ratner and G. Marom, J. Appl. Polym. Sci., 2002, 83, 2873 CrossRef CAS PubMed.
- H. Cai, F. Yan and Q. Xue, Mater. Sci. Eng., A, 2004, 364, 94 CrossRef.
- H. H. So, J. W. Cho and N. G. Sahoo, Eur. Polym. J., 2007, 43, 3750 CrossRef CAS PubMed.
- B.-K. Zhu, S.-H. Xie, Z.-K. Xu and Y.-Y. Xu, Compos. Sci. Technol., 2006, 66, 548 CrossRef CAS PubMed.
- X. Jiang, Y. Bin and M. Matsuo, Polymer, 2005, 46, 7418 CrossRef CAS PubMed.
- G. Xu, Y. Zhuang, R. Xia, J. Cheng and Y. Zhang, Mater. Lett., 2012, 89, 272 CrossRef CAS PubMed.
- E. D. Laird and C. Y. Li, Macromolecules, 2013, 46, 2877 CrossRef CAS.
- L. Li, Y. Yang, G. Yang, X. Chen, B. S. Hsiao, B. Chu, J. E. Spanier and C. Y. Li, Nano Lett., 2006, 6, 1007 CrossRef CAS PubMed.
- L. Li, C. Y. Li and C. Ni, J. Am. Chem. Soc., 2006, 128, 1692 CrossRef CAS PubMed.
- E. Zhuravlev, A. Wurm, P. Pötschke, R. Androsch, J. W. P. Schmelzer and C. Schick, Eur. Polym. J., 2014, 52, 1 CrossRef CAS PubMed.
- G. Z. Papageorgiou, M. Nerantzaki, I. Grigoriadou, D. G. Papageorgiou, K. Chrissafis and D. Bikiaris, Macromol. Chem. Phys., 2013, 214, 2415 CrossRef CAS PubMed.
- B. P. Grady, F. Pompeo, R. L. Shambaugh and D. E. Resasco, J. Phys. Chem. B, 2002, 106, 5852 CrossRef CAS.
- A. R. Bhattacharyya, T. Sreekumar, T. Liu, S. Kumar, L. M. Ericson, R. H. Hauge and R. E. Smalley, Polymer, 2003, 44, 2373 CrossRef CAS.
- M. Minus, H. G. Chae and S. Kumar, Polymer, 2006, 47, 3705 CrossRef CAS PubMed.
- Y.-Y. Liang, J.-Z. Xu, X.-Y. Liu, G.-J. Zhong and Z.-M. Li, Polymer, 2013, 54, 6479 CrossRef CAS PubMed.
- R. Haggenmueller, J. E. Fischer and K. I. Winey, Macromolecules, 2006, 39, 2964 CrossRef CAS.
- A. M. Díez-Pascual, M. Naffakh, M. A. Gómez, C. Marco, G. Ellis, M. T. Martínez, A. Ansón, J. M. González-Domínguez, Y. Martínez-Rubi and B. Simard, Carbon, 2009, 47, 3079 CrossRef PubMed.
- A. M. Díez-Pascual, M. Naffakh, C. Marco, G. Ellis and M. A. Gómez-Fatou, Prog. Mater. Sci., 2012, 57, 1106 CrossRef PubMed.
- H. G. Chae, M. L. Minus and S. Kumar, Polymer, 2006, 47, 3494 CrossRef CAS PubMed.
- G. O'Bryan, E. L. Yang, T. Zifer, K. Wally, J. L. Skinner and A. L. Vance, J. Appl. Polym. Sci., 2011, 120, 1379 CrossRef PubMed.
- Y. Dias and R. Yerushalmi-Rozen, Polymer, 2013, 54, 6399 CrossRef CAS PubMed.
- J. Kaur, J. H. Lee and M. L. Shofner, Polymer, 2011, 52, 4337 CrossRef CAS PubMed.
- X. L. Xie, Y. W. Mai and X. Ping, Mater. Sci. Eng., R, 2005, 49, 89 CrossRef PubMed.
- D.-H. Jung, Y. K. Ko and H.-T. Jung, Mater. Sci. Eng., C, 2004, 24, 117 CrossRef PubMed.
- L. Vaisman, G. Marom and H. D. Wagner, Adv. Funct. Mater., 2006, 16, 357 CrossRef CAS PubMed.
- A. Eitan, K. Jiang, D. Dukes, R. Andrews and L. S. Schadler, Chem. Mater., 2003, 15, 3198 CrossRef CAS.
- P. M. Ajayan and J. M. Tour, Nature, 2007, 447, 1066 CrossRef CAS PubMed.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS PubMed.
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzingger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760 CrossRef CAS PubMed.
- S. Goyanes, G. R. Rubiolo, A. Salazar, A. Jimeno, M. A. Corcuera and I. Mondragon, Diamond Relat. Mater., 2007, 16, 412 CrossRef CAS PubMed.
- M. A. Hamon, H. Hu, P. Bhowmik, S. Niyogi, B. Zhao, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2001, 347, 8 CrossRef CAS.
- K. Jiang, N. Grobert, A. Eitan, L. S. Schadler, P. M. Ajayan, R. W. Siegel, N. Grobert, M. Mayne, M. Reyes-Reyes, H. Terrones and M. Terrones, Nano Lett., 2003, 3, 275 CrossRef CAS.
- A. Eitan, K. Jiang, D. Dukes, R. Andrews and L. S. Schadler, Chem. Mater., 2003, 15, 3198 CrossRef CAS.
- S. V. Larin, S. G. Falkovich, V. M. Nazarychev, A. A. Gurtovenko, A. V. Lyulin and S. V. Lyulin, RSC Adv., 2014, 4, 830 RSC.
- S. G. Falkovich, S. V. Larin, A. V. Lyulin, V. E. Yudin, J. M. Kenny and S. V. Lyulin, RSC Adv., 2014, 4, 48606 RSC.
- G. Allegra, G. Raos and M. Vacatello, Prog. Polym. Sci., 2008, 33, 683 CrossRef CAS PubMed.
- Q. H. Zheng, A. B. Yu and G. Q. Lu, Prog. Polym. Sci., 2008, 33, 191 CrossRef PubMed.
- P. V. Komarov, Y.-T. Chiu, S.-M. Chen and P. Reineker, Macromol. Theory Simul., 2010, 19, 64 CAS.
- V. A. Ivanov, A. S. Rodionova, J. A. Martemyanova, M. R. Stukan, M. Muller, W. Paul and K. Binder, Macromolecules, 2014, 47, 1206 CrossRef CAS; M. Anwar, J. T. Berryman and T. Schilling, J. Chem. Phys., 2014, 141, 9 CrossRef PubMed; M. Anwar, F. Turci and T. Schilling, J. Chem. Phys., 2013, 139, 214904 CrossRef PubMed.
- S. V. Lyulin, S. V. Larin, A. A. Gurtovenko, V. M. Nazarychev, S. G. Falkovich, V. E. Yudin, V. M. Svetlichnyi, I. V. Gofman and A. V. Lyulin, Soft Matter, 2014, 10, 1224 RSC.
- V. Yudin, G. Divoux, J. Otaigbe and V. Svetlichnyi, Polymer, 2005, 46, 10866 CrossRef CAS PubMed.
- S. V. Lyulin, S. V. Larin, A. A. Gurtovenko, N. V. Lukasheva, V. E. Yudin, V. M. Svetlichyi and A. V. Lyulin, Polym. Sci., Ser. A, 2012, 54, 631 CrossRef CAS.
- V. M. Nazarychev, S. V. Larin, N. V. Lukasheva, A. D. Glova and S. V. Lyulin, Polym. Sci., Ser. A, 2013, 55, 570 CrossRef CAS.
- S. V. Lyulin, A. A. Gurtovenko, S. V. Larin, V. M. Nazarychev and A. V. Lyulin, Macromolecules, 2013, 46, 6357 CrossRef CAS.
- S. Falkovich, S. Lyulin, V. Nazarychev, S. Larin, A. Gurtovenko, N. Lukasheva and A. Lyulin, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 640 CrossRef CAS PubMed.
- S. Falkovich, S. Larin, V. Nazarychev, I. Volgin, A. Gurtovenko, A. Lyulin and S. Lyulin, Polym. Sci., Ser. A, 2014, 56, 558 CrossRef CAS.
- V. Nazarychev, S. Larin, A. Yakimanskii, N. Lukasheva, A. Gurtovenko, I. Gofman, V. Yudin, V. Svetlichyi, J. Kenny and S. Lyulin, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 912 CrossRef CAS PubMed.
- Y. Wang, L. Y. Jiang, T. Matsuurac, T. S. Chung and S. H. Goh, J. Membr. Sci., 2008, 318, 217 CrossRef CAS PubMed.
- N. Peng, T. S. Chung, M. L. Chung and W. J. Aw, J. Membr. Sci., 2010, 360, 48 CrossRef CAS PubMed.
- J. Xia, S. Liu, P. K. Pallathadka, M. L. Chng and T. Chung, Ind. Eng. Chem. Res., 2010, 49, 12014 CrossRef CAS.
- Polyetherimide resin additives, http://www.sabic-ip.com/gep/en/ProductsAndServices/SpecialtyAdditivesandIntermediatesProductPages/ultempeiresinsadditives.html, (accessed April 2015).
- Polyetherimide resin additives, http://www.sabic-ip.com/gep/en/ProductsAndServices/SpecialtyAdditivesandIntermediatesProductPages/extemthermoplasticpolyimideresins.html, (accessed April 2015).
- B. Hess, C. Kutzner, D. Van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435 CrossRef CAS; D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhoff, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701 CrossRef PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656 CrossRef CAS PubMed.
- J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Di Nola and J. R. J. Haak, Chem. Phys., 1984, 81, 3684 Search PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463 CrossRef CAS.
- C. Oostenbrink, T. A. Soares, N. F. A. Van der Vegt and W. F. Van Gunsteren, Eur. Biophys. J., 2005, 34, 273 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577 CrossRef CAS PubMed.
- A. Karantrasos, R. J. Composto, K. I. Winey and N. Clarke, Macromolecules, 2011, 44, 9830 CrossRef.
- S. Chakraborty and S. Roy, J. Phys. Chem. B, 2012, 116, 3083 CrossRef CAS PubMed.
- D. Qi, J. Hinkley and G. He, Modell. Simul. Mater. Sci. Eng., 2005, 13, 493 CrossRef CAS.
- R. Rahman and A. Haque, Composites, Part B, 2013, 54, 353 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |