
Anti-site disorder and physical properties in microwave synthesized RE2Ti2O7 (RE = Gd, Ho) pyrochlores
Alejandro Gómez-Péreza,
Jesús Prado-Gonjalb,
Daniel Muñoz-Gilb,
Adrián Andrada-Chacónc,
Javier Sánchez-Benítezc,
Emilio Moránb,
María Teresa Azcondoa,
Ulises Amadora and
Rainer Schmidt*d
aDpto. Química, Facultad de Farmacia, Universidad San Pablo – CEU, 28668 Boadilla del Monte, Madrid, Spain
bDpto. Química Inorgánica, Facultad de CC. Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain
cDpto. Química Física I, Facultad de CC. Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain
dDpto. Física Aplicada III, Facultad de CC. Físicas, GFMC, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: rainerxschmidt@googlemail.com
First published on 2nd October 2015
Abstract
In this work we report on the microwave assisted synthesis of nano-sized Gd2Ti2O7 (GTO) and Ho2Ti2O7 (HTO) powders from the RE2Ti2O7 pyrochlore family (RE = rare earth). Synchrotron X-ray powder diffraction was used to study RE–Ti cationic anti-site defects with concentrations that decrease in both samples with increasing temperature starting from 1100 °C, and the defects disappear at 1400 °C. SQUID magnetometry measurements revealed that GTO shows a predominantly anti-ferromagnetic structure, whereas HTO exhibits magnetic saturation and a ferromagnetic component at low temperature. Impedance spectroscopy data revealed strongly increased ionic oxygen vacancy conduction in HTO ceramic pellets as compared to GTO, which may be associated with a higher degree of oxygen vacancy disorder. This argument was supported by Raman spectroscopy data.
1. Introduction
Pyrochlore oxide compounds of the type A2B2O7 are functional materials for a wide range of potential applications due to their high oxygen ion conductivity, dielectric tunability, exotic magnetic properties, multiferroic behaviour and superconductivity.1–7 Furthermore, due to their high chemical stability and flexibility to form solid solutions with many cations, pyrochlores have been proposed as a host matrix for immobilizing toxic nuclear waste.8,9Rare-Earth (RE) titanate pyrochlores have the general formula RE2Ti2O7, where the crystal structure may be regarded a RE3+ and Ti4+ cation ordered superstructure of oxygen deficient fluorite (RE0.5Ti0.5O2-0.25). A concentration of 0.25 oxygen vacancies per formula unit allows ionic oxygen vacancy conduction to occur. The pyrochlore structure (Fig. 1) exhibits an F-centred cubic unit cell with space group (S.G): Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (#227) and cell parameter a ≈ 10 Å, which corresponds to approximately the double of the fluorite unit cell parameter.10 The larger RE3+ cations are located on 16d eight-coordinated sites at the centre of scalenohedrons, while Ti4+ are located on 16c six-coordinated sites at the centre of trigonal antiprisms. The 7 oxygen O2− anions in the chemical formula RE2Ti2O6O′ are located on two different sites: (1) 6 oxygen O(1) on four-coordinated 48f positions with two A cation and two B cation nearest neighbours, and (2) one oxygen O(2) on four-coordinated 8b sites with four A cation nearest neighbours. The third anionic tetrahedral O(3) sites are empty, consisting in four-coordinated 8a sites surrounded by four B cation neighbours. These systematically vacant O(3) sites are characteristic of the ordered pyrochlore structure, in contrast to anion deficient fluorites where anion vacancies are randomly distributed throughout the anion sub-structure. Merely based on steric considerations regarding the cationic radii r, one would expect the eight-coordinated 16d sites in RE2Ti2O7 pyrochlores to be fully occupied by the larger RE3+ cations, whereas the smaller Ti4+ would be located on the six-coordinated 16c positions: r(VIIIGd3+) = 1.053 Å, r(VIIIHo3+) = 1.015 Å and r(VITi4+) = 0.605 Å.11 However, in the A2B2O7 pyrochlore structure crystal defects commonly occur, where the two most important ones are (i) cation anti-site, and (ii) anion Frenkel defects. Cation anti-site defects consist in a positional exchange of an A3+ (here RE3+) with a B4+ (here Ti4+) cation, whereas Frenkel anion disorder occurs when a 48f site oxygen anion is displaced to a nominally vacant 8a interstitial site, thus leaving a 48f site unoccupied.10,12
m (#227) and cell parameter a ≈ 10 Å, which corresponds to approximately the double of the fluorite unit cell parameter.10 The larger RE3+ cations are located on 16d eight-coordinated sites at the centre of scalenohedrons, while Ti4+ are located on 16c six-coordinated sites at the centre of trigonal antiprisms. The 7 oxygen O2− anions in the chemical formula RE2Ti2O6O′ are located on two different sites: (1) 6 oxygen O(1) on four-coordinated 48f positions with two A cation and two B cation nearest neighbours, and (2) one oxygen O(2) on four-coordinated 8b sites with four A cation nearest neighbours. The third anionic tetrahedral O(3) sites are empty, consisting in four-coordinated 8a sites surrounded by four B cation neighbours. These systematically vacant O(3) sites are characteristic of the ordered pyrochlore structure, in contrast to anion deficient fluorites where anion vacancies are randomly distributed throughout the anion sub-structure. Merely based on steric considerations regarding the cationic radii r, one would expect the eight-coordinated 16d sites in RE2Ti2O7 pyrochlores to be fully occupied by the larger RE3+ cations, whereas the smaller Ti4+ would be located on the six-coordinated 16c positions: r(VIIIGd3+) = 1.053 Å, r(VIIIHo3+) = 1.015 Å and r(VITi4+) = 0.605 Å.11 However, in the A2B2O7 pyrochlore structure crystal defects commonly occur, where the two most important ones are (i) cation anti-site, and (ii) anion Frenkel defects. Cation anti-site defects consist in a positional exchange of an A3+ (here RE3+) with a B4+ (here Ti4+) cation, whereas Frenkel anion disorder occurs when a 48f site oxygen anion is displaced to a nominally vacant 8a interstitial site, thus leaving a 48f site unoccupied.10,12
 | ||
| Fig. 1 Schematic representation of the ideal RE2Ti2O7 pyrochlore structure. Trigonal antiprisms (green) contain the 16c six-coordinated Ti4+ cations at the centre and the 48f oxygen O(1) at the corners (not shown). The RE3+ cations (green) are located at the centre of a scalenohedron (not drawn out), where 8 of the 10 corners are occupied by the 48f O(1) and the 8b O(2) oxygen, the latter are shown in red. | ||
Disordered oxygen vacancies (e.g. in oxygen deficient fluorites) favour higher oxygen ion mobility and conductivity.13–16 Therefore, high oxygen mobility and conductivity in the pyrochlore structure was claimed to be favoured by a certain degree of A and B anti-site defects, because anti-site defects may be directly coupled to the oxygen vacancy disorder, i.e. increased anti-site defects directly induce increased oxygen vacancy disorder in the nominally oxygen ordered structure and improve oxygen ion conductivity.13–15 In this context it should be noted that increasing concentrations of pyrochlore A and B anti-site defects can be regarded a precursor to the formation of the fluorite structure with a disordered arrangement of the cationic sub-lattice and the oxygen vacancies. On the other hand, some authors have reported that some degree of oxygen structural disorder can exists in pyrochlores despite a fully ordered cationic sub-lattice, as evidenced by Raman spectroscopy, and the anti-site and anion Frenkel defects may not always be directly coupled.17,18
Here in this work the coupling of anti-site and Frenkel defects could not be confirmed for the HTO samples, because a negligible anti-site defect concentration was detected by synchrotron X-ray diffraction (SXRD), but Raman spectroscopy suggested considerable oxygen vacancy disorder in HTO. The latter finding was corroborated by significantly enhanced ionic oxygen vacancy conduction in the HTO sample as compared to GTO, detected by impedance spectroscopy on sintered ceramic pellets.
It has been established previously that the concentration of pyrochlore A and B anti-site defects depends on the details of the synthesis procedure.17–19 Here in this work we demonstrate that the microwave (MW)-assisted method produces off-equilibrium (metastable) pyrochlore-type phases with varying amounts of anti-site defects, in agreement with other more recently developed alternative synthesis methods, e.g. mechanical milling.19 Traditionally, RE2Ti2O7 compounds are prepared by solid state reactions, which require repeated and long synthesis procedures at temperatures of up to ≈1400 °C and intensive grinding at the initial and intermediate stages of processing to achieve single phase material.20–22 In the last decades several alternative routes have been proposed such as microwave (MW) synthesis, molten salt mediated synthesis, co-precipitation of hydroxides, sol–gel and citrate based methods, and mechanical milling, where the time and temperature requirements for synthesis are significantly reduced.19,23–26 However, these methods usually require a second calcination step in a conventional furnace to obtain phase pure materials and the total reaction time increases. MW-assisted synthesis may be particularly useful due to the nanometre-sized particles that can be obtained at reduced synthesis time and processing costs.27–32 In the work presented here we use a domestic microwave oven for the first heat treatment process of only 20 min, followed by a 2 h calcination procedure at 1100 °C in a conventional furnace to obtain highly crystalline single phase materials.33 We explore this fast MW-assisted synthetic route for RE2Ti2O7 pyrochlores with RE = Gd, Ho. The nano-metric particle sizes may improve the sintering activity of the resulting powders, which is beneficial to obtain dense bulk materials by ceramic sintering to study the dielectric properties. Furthermore, a quick and economic sintering process may be relevant for potential industrial applications. By example, we present the microwave synthesis of Gd2Ti2O7 (GTO) and Ho2Ti2O7 (HTO) pyrochlores, and their structural, magnetic and dielectric properties.
2. Experimental
2.1. Synthesis
Highly reactive TiO2 amorphous powder was previously obtained from titanium(IV) propoxide dissolved in isopropyl alcohol, and the mixture was hydrolysed by adding water. An amorphous bulky gel was obtained, which was filtered and dried at 80 °C for 12 hours. Furthermore, the respective RE-nitrates (RE(NO3)3·xH2O) (99.9% Sigma-Aldrich) were used as precursor oxides, where the amounts of water of hydration (x = 4.61 for RE = Gd and x = 3.54 for RE = Ho) were determined previously by thermogravimetric analysis. For one sample batch ≈1.3 g of the stoichiometric precursor mixture was homogenized together with 5% (wt) carbon black powder, which serves as a microwave absorber to improve the heat transfer. The mixture was ground intimately and pressed into pellets of 13 mm diameter and ≈650 mg using a uniaxial press, and the pellets were placed into a small porcelain crucible inside another larger one stuffed with mullite. The crucibles were MW-irradiated in a domestic microwave oven operating at 2.45 GHz frequency and a power of 800 W. After MW irradiation for 20 min, in a second heating step the pellets were calcined at 1100 °C for 2 h using a conventional furnace. The 2 h holding time and 1100 °C temperature were determined by a trial-and-error procedure, where calcining at 800 °C for 12 h and at 1000 °C for 12 h both proved to be insufficient to obtain a single pyrochlore phase. Furthermore, a control experiment was performed to synthesize GTO powder at 1200 °C for 3 h without the previous microwave heating step, but again no single phase could be obtained. Additional control experiments were carried out to prove the reproducibility of the synthesis process by using different microwave furnaces under the same conditions. All together, we have used 3 different domestic microwave furnaces (Sanyo EM – S1057G ES2; Balay 3WMX1918X 17L; CATA FS 20 WH) all operating at 2.45 GHz frequency and 800 W. After the second calcination step the resulting phases were confirmed to be all reproducible by standard X-ray diffraction (XRD).In the field of solid state synthesis especially domestic microwave furnaces are well-known to often entail problems with the reproducibility of the final products due to the lack of temperature control. In this work we did not encounter this problem, most likely due to the fact that we used a microwave heating source only for the first synthesis step to produce precursor powder followed by a second step calcination process in a conventional furnace. In the conventional furnace the temperature is well-controlled and reproducibility problems usually do not occur.
All samples were analysed by standard XRD and scanning electron microscopy (SEM) twice: first, after the initial microwave treatment and second, after the subsequent calcination treatment at 1100 °C in the conventional furnace. In each case the pellets were extracted from the microwave or the conventional furnaces and were ground into powder form. Three batches of the final single phase products were further heated at 1200 °C, 1300 °C and 1400 °C for 12 h each to study the temperature dependence of the concentration of anti-site defects. For ionic conductivity measurements by impedance spectroscopy, GTO and HTO pressed pellets of 5 mm diameter and 2 mm thickness were sintered at 1400 °C for 4 h (heating and cooling ramps of 2 °C min−1). The relative densities of the sintered GTO and HTO pellets were ≈95% of the crystallographic value. Both faces of the pellets were covered with Au electrodes using dc sputtering to facilitate impedance spectroscopy measurements.
2.2. Characterization
The phase composition of all samples were determined by conventional X-ray diffraction (XRD) on a PANAnalytical X'Pert PRO MPD apparatus using CuKα radiation.High-resolution powder synchrotron X-ray diffraction (SXRD) patterns were collected at the SpLine beamline (BM25A) of the Spanish CRG at the European Synchrotron Radiation Facility (ESRF, Grenoble) with a fixed wavelength of λ = 0.49684897 Å (24954.112 eV). Powdered samples were placed inside capillaries with diameters ranging from 0.2–0.5 mm, where the capillaries were rotated during exposure. In each case the absorption was calculated to maximize the signal-to-time ratio for every sample. Data were collected in a continuous 2θ-scan mode from 3° to 35°.
Structural refinements using (S)XRD data were carried out by the Rietveld method using FULLPROF software.34 A Pseudo-Voigt function was selected as the line profile and the corresponding parameters were refined. Furthermore, the set of pre-defined background points, scale factor, zero shift, lattice parameters, atomic O displacement and the overall Debye–Weller factors were refined. These overall factors were converted into individual isotropic Debye–Waller factors and refined. The cation occupation was also refined to detect possible anti-site defects.
Scanning electron microscopy (SEM) micrographs were collected on an FEI XL30® apparatus equipped with an attached analyser for energy dispersive spectroscopy (EDS).
Unpolarised Raman spectroscopy experiments were performed using a micro-Raman confocal spectrometer (Voyage™ BWS435-532SY, BW&Tek). An excitation wavelength of 532.0 nm was produced by a solid state laser with an incident power fixed at 10 mW. The spectrometer was equipped with an Olympus BX51 microscope allowing the collection of the Raman signal on backscattering configuration through a 50× long working distance objective. With this setup, the spectral resolution was about 4 cm−1. A Chameleon™ CMLN-13S2C digital camera has also been coupled to the equipment.
Magnetic susceptibility measurements of GTO and HTO powder samples calcined at 1100 °C were performed in a Quantum Design XL-SQUID magnetometer in the temperature range of 2–300 K at 1 kOe applied magnetic field. The temperature dependence of the magnetic susceptibility was measured following zero-field-cooled and field-cooled (ZFC–FC) procedures with intermediate demagnetization at room temperature. Ferri-magnetic hysteresis cycles were recorded at 1.7 K from magnetization vs. applied field measurements.
Impedance spectroscopy was carried out on 1400 °C sintered ceramic pellets between 150 and 550 K using a Novocontrol Alpha-A High Performance Frequency Analyzer equipped with a nitrogen gas cooled sample chamber. A 100 mV amplitude alternating voltage signal was employed and the data was recorded between 0.5 Hz to 3 MHz in terms of the real and imaginary parts of the impedance (Z′–Z′′). The data were converted into the permittivity notation ε′–ε′′ using the standard conversion.35 High temperature impedance spectroscopy was carried out using a Solartron 1260 Frequency Response Analyzer in the 1 Hz to 1 MHz range between 298 and 1273 K in different atmospheres (air, pure oxygen or pure Ar), using a 100 mV amplitude alternating voltage signal.
3. Results and discussion
3.1. Reaction pathways
Fig. 2 shows the conventional XRD patterns from the 20 min MW-irradiated precursor mixtures for the nominal GTO compound after the subsequent calcination at 800 °C (Fig. 2a). Calcining at 800 °C for 12 h proved to be suitable to remove the traces from the carbon black powder used as a microwave absorber, but the synthesis reaction was still not complete and a mixture of GTO and the unreacted precursors Gd2O3 and TiO2 (anatase) is evident. The XRD pattern for the sample calcined at 1000 °C during 12 h (Fig. 2b) reveals that even at this elevated temperature some unreacted Gd2O3 and TiO2 (rutile) still remain. A better result was obtained after calcining at 1100 °C for only 2 h (Fig. 3a) and a single pyrochlore phase with high crystallinity is evident. A similar behaviour is observed for the Ho-containing sample. Fig. 3a and b demonstrate that both GTO and HTO powders can be obtained as pure single phases by calcining the 20 min MW-treated powders at 1100 °C for 2 h. Thus, it seems that the key parameter is the temperature rather than the holding time to complete the synthesis reaction. On the contrary, the XRD pattern of a sample of GTO prepared by the conventional calcination treatment (without previous microwave irradiation) (Fig. 3c) reveals that the reaction is not complete in spite of using higher temperature and larger reaction times (1200 °C, 3 hours). This is not surprising since the preparation of pyrochlore oxides by traditional solid-state reaction from a mixture of oxides commonly requires repeated grinding and long heating cycles at temperatures higher than 1200 °C to achieve complete reaction.20–22 Thus, we can confirm that the use of microwave heating is useful to reduce the reaction time, temperature and costs for the synthesis of RE-titanate pyrochlores. | ||
| Fig. 2 XRD pattern for Gd2Ti2O7 powders irradiated by microwaves for 20 min and subsequently calcined at 800 °C (a) and 1000 °C (b) for 12 h. Besides the pyrochlore material (first row of vertical bars) unreacted Gd2O3 (second row) and TiO2 (anatase in (a), rutile in (b), third row) are observed. | ||
 | ||
| Fig. 3 XRD pattern for (a) Gd2Ti2O7 and (b) Ho2Ti2O7, obtained from MW-irradiated precursors calcined at 1100 °C for 2 h and (c) for a non-MW-irradiated Gd2Ti2O7 sample calcined at 1200 °C for 3 h. In (a) and (b) only the pyrochlore phase is observed, in (c) pyrochlore (first row of vertical bars), unreacted Gd2O3 (second row) and TiO2 (rutile, third row) are observed. | ||
3.2. Microstructural characterization
Fig. 4 shows the backscattered-electron mode SEM micrographs of the single phase RE2Ti2O7 final product powders after 20 min of microwave irradiation and 2 h calcination at 1100 °C. The powders are composed of agglomerations of submicron or nanometric spherical particles with high homogeneity and a sponge-like texture. This morphology may arise most likely from the large amount of gases produced during the decomposition of the starting nitrates and oxidation of active carbon to CO2. It is worth mentioning that the small particle size persists after the second 1100 °C calcination step, facilitating a good sintering activity of the powders. Therefore, a short sintering process of the pressed pellets for 4 h at 1400 °C is sufficient to obtain highly densified ceramics as a result of significant and rapid particle growth, as demonstrated in Fig. 4c and d. EDS analyses confirmed the nominal metallic compositions (≈RE 50% and Ti 50%). Furthermore, the homogeneous contrast in the backscattered SEM images in Fig. 4 implies that RE and Ti cations are well dispersed across the samples discarding any segregation of impurity phases. | ||
| Fig. 4 SEM micrographs of the RE2Ti2O7 samples. (a) and (b) correspond to Gd2Ti2O7 and Ho2Ti2O7 powder samples. (c) and (d) correspond to the respective pellets sintered at 1400 °C during 4 h. | ||
3.3. Analysis of anti-site defects by SXRD
As mentioned above (Section 1), the pyrochlore structure can be considered a superstructure of an anion-deficient fluorite atomic arrangement. Therefore, the pyrochlore diffraction pattern contains a set of strong peaks characteristic of the fluorite substructure plus an additional set of superstructure reflections. The intensities of the latter depend on factors like the degree of ordering, difference in the average scattering factors of the elements involved and distribution of oxygen vacancies.13 Superstructure peaks due to the pyrochlore long-range atomic ordering are evident in the SXRD patterns from the GTO and HTO samples treated at different temperatures from 1100 °C to 1400 °C (Fig. 5 and 6). Since the temperature for the pellet sintering process was 1400 °C, the structural results obtained from the 1400 °C heated samples may also be representative for the sintered pellets, which were analysed by impedance spectroscopy to determine the ionic conductivities (Section 3.6). | ||
| Fig. 5 Rietveld refinement of synchrotron XRD patterns of Ho2Ti2O7 treated at 1100 °C and 1200 °C. Experimental pattern (red dotted line), calculated (black solid line), and their difference (blue bottom line) are depicted. Green vertical bars indicate the reflection positions. | ||
 | ||
| Fig. 6 Rietveld refinement of synchrotron XRD patterns of Gd2Ti2O7 treated at 1300 °C and 1400 °C. Experimental pattern (red dotted line), calculated (black solid line), and their difference (blue bottom line) are depicted. Green vertical bars indicate the reflection positions. | ||
The structure factors (Fhkl) of the superstructure peaks depend on the difference of scattering power between the 16d and the 16c sites and the Fhkl factors for peaks with even h, k and l depend on the total of the scattering power in both positions. Therefore, the relative intensities of the superstructure peaks are a direct reflection of the cation distribution in the structure.
We started our structural refinements of the SXRD data with the fully ordered pyrochlore structure, where all RE ions are constrained to the 16d sites and the Ti to the 16c sites. However, it became evident from the fitting process that more scattering power should be located in the 16c site which could only be accomplished by allowing the exchange of RE and Ti cations from their nominal positions, i.e. the occurrence of A and B anti-site defects. The final refined cation distributions presented in Table 1 show a perceptible fraction of Gd and Ho cations to be located at the six-coordinated 16c site, which reduced after heating the powders at higher temperatures (up to 1400 °C). The anti-site defect percentages are 3.6% for Gd2Ti2O7 and 2.4% for Ho2Ti2O7 at 1100 °C. This is consistent with the different relative intensities of the superstructure peaks shown in Fig. 7, where the anti-site defect concentrations show clear temperature dependence. Generally, anti-site defects are more common when the A and B cations have similar ionic radii, whereas in the RE2Ti2O7 case presented here the A- and B-site cationic radii are quite different and the anti-site defect concentration is then expected to be strongly temperature dependent.18,19,36 The temperature dependence of the anti-site defect concentrations is summarized in Table 1.
| Ho2Ti2O7 | Gd2Ti2O7 | ||||||
|---|---|---|---|---|---|---|---|
| 1100 °C | 1200 °C | 1400 °C | 1100 °C | 1200 °C | 1300 °C | 1400 °C | |
| a (Å) | 10.10004(6) | 10.09882(5) | 10.09828(3) | 10.18126(8) | 10.18407(5) | 10.18496(5) | 10.18507(6) |
| A position | 16d | 16d | |||||
| U × 100 (Å2) | 0.72(1) | 0.74(2) | 0.38(3) | 0.84(1) | 0.62(2) | 0.56(3) | 0.45(2) |
| Occ RE/Ti | 1.952(3)/0.048(3) | 1.952(2)/0.048(2) | 2.000(2)/0.000(2) | 1.929(3)/0.071(3) | 1.952(2)/0.048(2) | 1.976(2)/0.024(2) | 2.000(2)/0.000(2) |
| B position | 16c | 16c | |||||
| U × 100 (Å2) | 1.01(5) | 1.03(3) | 0.39(2) | 1.22(2) | 0.83(1) | 0.65(2) | 0.95(4) |
| Occ Ti/RE | 1.952(3)/0.048(3) | 1.952(2)/0.048(2) | 2.000(2)/0.000(2) | 1.929(3)/0.071(3) | 1.952(2)/0.048(2) | 1.976(2)/0.024(2) | 2.000(2)/0.000(2) |
| O(1) position | 48f | 48f | |||||
| X | 0.4229(4) | 0.4232(4) | 0.4227(4) | 0.4239(4) | 0.4244(3) | 0.4253(3) | 0.42877(5) |
| U × 100 (Å2) | 0.71(2) | 0.71(2) | 0.42(1) | 0.82(3) | 1.13(1) | 0.88(2) | 0.93(1) |
| O(2) position | 8b | 8b | |||||
| U × 100 (Å2) | 0.09(2) | 0.02(4) | 0.51(3) | 0.09(2) | 0.16(3) | 0.01(2) | 0.23(3) |
| χ2 | 4.24 | 4.43 | 4.60 | 4.78 | 4.74 | 3.57 | 6.21 |
| Rwp/Rexp (%/%) | 9.63/4.67 | 9.13/4.34 | 9.36/4.36 | 10.7/4.92 | 9.23/4.24 | 7.59/4.02 | 12.3/3.94 |
| RBragg | 2.42 | 2.35 | 2.83 | 2.40 | 2.16 | 1.90 | 5.50 |
| Space group | Fdm (#227): 16d (1/2 1/2 1/2), 16c (0 0 0), 48f (x 1/8 1/8), 4b (3/8 3/8 3/8) |
Fdm (#227): 16d (1/2 1/2 1/2), 16c (0 0 0), 48f (x 1/8 1/8), 4b (3/8 3/8 3/8) |
|||||
 | ||
| Fig. 7 Left axis: Temperature dependence of the intensity of the superstructure peaks (111) in red and (331) in blue, referred to that of the (222) peak for GTO (solid lines) and HTO (dashed lines). Right axis: Anti-site (AS)-defect concentrations for Gd2Ti2O7 (solid squares) and Ho2Ti2O7 (asterisks). Lines are a guide for the eyes. | ||
The pyrochlore unit cell parameter is mainly determined by the size of the BO6 distorted octahedra (=trigonal antiprisms) and, bearing in mind the ionic radii of RE3+ and Ti4+ cations, it is clear that a higher degree of occupation of the 16c sites by RE cations increases the size of the BO6 units and concomitantly the unit cell.19 Since thermal treatment removes anti-site defects it would therefore be expected that heating at higher temperatures would result in smaller unit cell parameters. Additionally, the unit cell parameters may also be affected by a small concentration (δ) of non-stoichiometric oxygen vacancies (RE2Ti2O7−δ), compensated by the formation of Ti3+.37,38 By heating at high temperature in oxidizing conditions these anion vacancies would be filled, accompanied by oxidation of large Ti3+ cations to smaller Ti4+, and the unit cell parameter would again decrease. We believe that non-stoichiometric oxygen vacancies may well exist in our samples since we use strongly reducing carbon as MW-absorber and the obtained products would therefore be expected to be oxygen-deficient to a certain extent. Therefore, we presume that at least two mechanisms may operate at the same time during heat treatment at elevated temperatures in air: (i) cation re-distribution among A- and B-sites, and (ii) oxidation of Ti3+ to Ti4+ through incorporation of oxygen into non-stoichiometric anion vacancies. Both processes would induce shortening of the unit cell.
The unit cell size for HTO (Table 1) displays such a trend, whereas for GTO the unit cell shows an expansion as the temperature of the heat treatments increases. We suspect that the thermal expansion of the GTO lattice may dominate over the decreasing anti-site defect concentration and/or the concentration of non-stoichiometric oxygen vacancies may be significantly lower, which then results in a different evolution of the unit cell size. Alternatively, the GTO phase may be affected by additional structural variations.
3.4. Raman spectroscopy
Fig. 8 shows the Raman spectra for GTO and HTO samples heat treated at different temperatures. The observed Raman modes have been assigned to F2g and A1g modes according to the literature,39–42 where some unassigned modes (small black arrows) have been interpreted as two-phonon modes. The Raman modes in Fig. 8 have different intensities due to the different RE3+ cations in GTO and HTO, which is expected according to the literature. The drastic change of Raman mode intensities with the heat treatment temperature observed in the GTO sample (but not in the HTO) is unusual and may point towards additional structural variations. This could possibly be correlated with the unusual changes of the unit cell volume mentioned above in Section 3.3.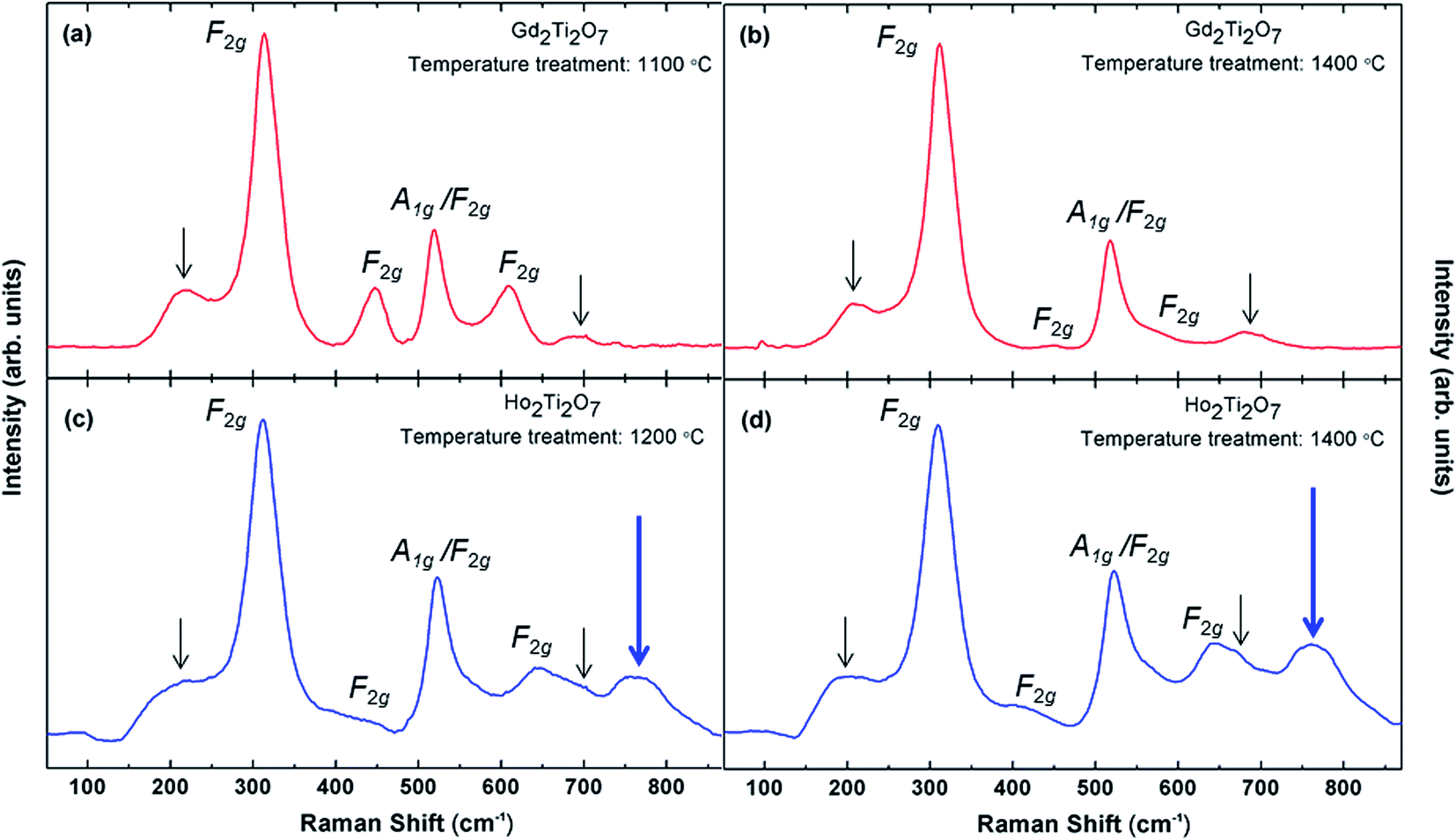 | ||
| Fig. 8 Raman shift. (a) Gd2Ti2O7 (red) heat treated at 1100 °C, (b) Gd2Ti2O7 heat treated at 1400 °C, (c) Ho2Ti2O7 (blue) heat treated at 1200 °C, (d) Ho2Ti2O7 heat treated at 1400 °C. | ||
The main feature of the Raman spectra shown in Fig. 8 is the appearance of an additional phonon (large blue arrows) centred at 750 cm−1 in HTO (but not in GTO). This mode has been observed previously in Y2Zr2−yTiyO7 pyrochlore structures and was associated with a substructure with varying oxygen coordination.42 We interpret the occurrence of this mode as a clear sign of oxygen disorder in the HTO, and the two types of defects, anti-site and Frenkel anion defects, may be uncoupled to a certain extent. This notion is corroborated by a strongly increased ionic oxygen vacancy conduction in the HTO sample as demonstrated below (Section 3.6) despite the absence of anti-site disorder detected by SXRD measurements (Section 3.3).
3.5. Magnetic properties of the magnetic structure
The magnetic structure of RE2Ti2O7 (RE = Gd, Ho) pyrochlores is determined by the magnetically active RE-cations, which form a tetrahedral sublattice where the tetrahedral antiferromagnetic interactions exhibit strong frustration.43 The magnetic ground state has been referred to as a spin ice, where a “two spins in, two spins out” configuration may be satisfied in every tetrahedron.44,45Fig. 9 shows the temperature dependence of the magnetic susceptibility for both powder samples. No significant differences between ZFC (solid marks) and FC (hollow marks) susceptibility curves are evident. The insets in Fig. 9 show plots of the reciprocal magnetic susceptibility 1/χ vs. T at low temperature, indicating a deviation from the Curie–Weiss law [χ = C/(T − θ)] below ≈10 K in both samples. The Curie–Weiss fits are indicated by black solid lines, which allowed the extraction of the Curie constant C, related to the effective magnetic moment μeff, and the Weiss constant θ. In the case of GTO a linear regression analysis yields μeff ≈ 5.7 μB/Gd3+ and θ ≈ −9.8 K. A negative θ value suggests that antiferromagnetic interactions may be predominant. For HTO, μeff ≈ 7.3 μB/Ho3+, and θ ≈ +0.2 K. The latter positive value suggests that ferromagnetic interactions may be predominant. The experimental magnetic moments are smaller than the theoretical ones: μeff = 7.9 μB for the free ion 8S7/2 ground state of Gd3+ and 10.6 μB for the 5I8 Ho3+ ground state. This discrepancy between theoretical and experimental magnetic moments may be related to the magnetic frustration commonly encountered in pyrochlore structures.
 | ||
| Fig. 9 Temperature dependence of the magnetic susceptibilities χ for Gd2Ti2O7 (red circles) and Ho2Ti2O7 (blue squares) at 1 kOe. Solid and empty symbols represent ZFC and FC data respectively. Insets: inverse susceptibility 1/χ. The black line represents the fit to the Curie–Weiss law. A deviation is demonstrated below ≈ 10 K. | ||
Fig. 10 presents the magnetization vs. applied field plots at 1.7 K for GTO and HTO, where a small hysteresis is observed. In the case of HTO a magnetic saturation is observed, which confirms the positive value of the Weiss constant and the notion that a soft ferromagnetic component drives the magnetic behaviour. A more detailed investigation into the magnetic structure of the GTO and HTO phases in terms of neutron diffraction experiments may be indicated to elucidate the origin of the ferromagnetic component in HTO, but this goes behind the scope of the work presented here.
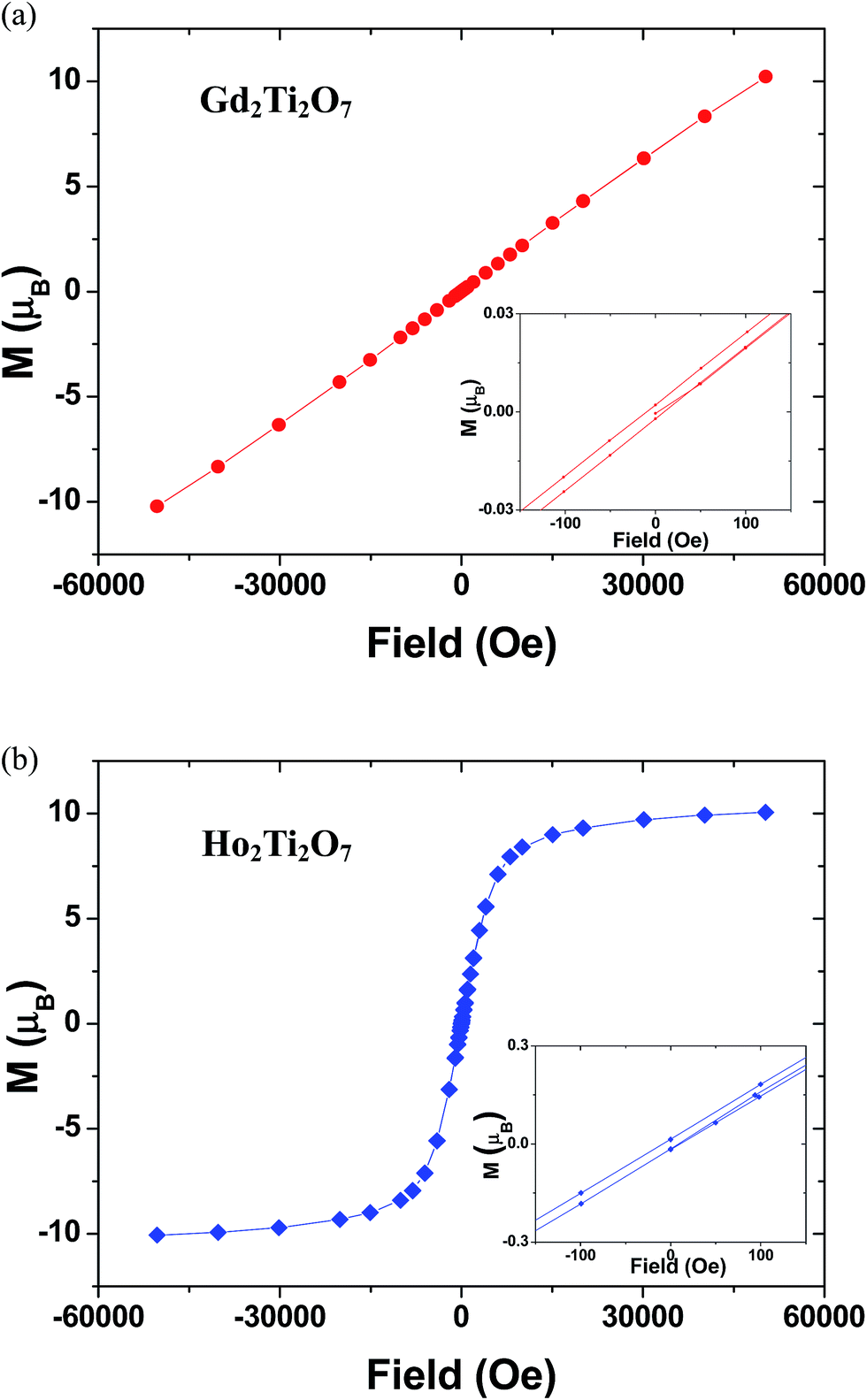 | ||
| Fig. 10 Magnetic hysteresis loop at 1.7 K in the magnetic field range of −50 to 50 kOe for (a) Gd2Ti2O7 (red circles) and (b) Ho2Ti2O7 (blue squares). Insets: magnification of the hysteresis loops. | ||
3.6. Dielectric properties
Impedance Spectroscopy (IS) data collected at 560 K for GTO and HTO ceramic pellets sintered at 1400 °C are shown in Fig. 11 in terms of the complex impedance plane plots of −Z′′ vs. Z′. For both samples (Fig. 11a and b) one regular semicircle is displayed. The data can therefore be modeled by a resistor–capacitor (RC) element where the resistance R corresponds to the respective semicircle diameter. The specific capacitance (discussed below) extracted from the single semicircles is in an order of magnitude that indicates that this dielectric contribution can be ascribed to an intrinsic grain interior bulk effect.46 | ||
| Fig. 11 Complex impedance plane plots of −Z′′ vs. Z′ for (a) Gd2Ti2O7 (red open diamonds) and (b) Ho2Ti2O7 (blue open diamonds) ceramic pellets. The equivalent circuits used for fitting the data are depicted in the figure insets. Solid black squares and lines indicate the fitted curves. | ||
At the low frequency end the data points for the HTO sample in the −Z′′ vs. Z′ notation (Fig. 11b) align linearly in a “pike-like” fashion, which is inconsistent with the conventional RC element model. This behavior is commonly interpreted as a blocking effect of the electrode sample interface (IF).35 This constitutes evidence for ionic charge transport where the blocking effect of the interface occurs as a result of the different types of dominating charge carriers in the Au electrodes (electrons) and in the ceramic (oxygen vacancies). Therefore, ionic oxygen vacancy conductivity in the HTO samples is indicated, and electronic contributions may be small. The charge transport across blocking electrode interfaces is usually by diffusion, which was not accounted for in the equivalent circuit model here, because rather complex circuit components such as Warburg elements are needed to describe diffusion processes by an equivalent circuit component.35 For the GTO sample the interface pike contribution is not visible in Fig. 11a, most likely due to insufficient resolution at the low frequency end. The interface contribution may possibly appear at lower frequency. This may be attributed to the higher resistivity of the GTO sample, which indicates a slower charge transport mechanism at the same temperature, and the same features in the impedance spectra would be visible at lower frequency.
The resulting equivalent circuits used for fitting the data from GTO and HTO ceramic pellets are displayed in the insets of Fig. 11a and b and a good fit to the data can be obtained in the high and intermediate frequency range where the bulk contributions are dominant. It should be noted that the semicircles in Fig. 11 are slightly suppressed in a way that the semicircle center seems to be slightly shifted below the Z′ x-axis. This indicates a certain degree of non-ideality of the dielectric relaxation processes, which can be accounted for by connecting a constant phase element (CPE) in parallel to the ideal RC element.47 The CPE behavior is usually explained in the framework of a broadening of the distribution of relaxation times τ across the macroscopic sample, where τ = RC, with R being the resistance and C the capacitance of an ideal RC element.35,48
In both cases, for GTO and HTO, only one semicircle is evident from Fig. 11, although the pike-like interface contribution suggests ionic type charge transport, at least in the HTO. This is rather unusual, because ionic charge transport is understood to be inhibited by Schottky-type charge transport barriers at the grain boundary (GB) regions, and both bulk and GB contributions are expected to appear in form of two separated semicircles in −Z′′ vs. Z′ plots.46 The reasons for the disappearance of the GB contributions are not entirely clear but the GB Schottky model may possibly not be valid here.
The ionic conductivity in the HTO sample is larger (the resistivity is lower) as compared to GTO, which is obvious from the different semicircle diameter (the difference is about a factor of 150). This large difference in resistivity is also evident in Fig. 12, where semi-logarithmic plots of resistivity ρ vs. reciprocal temperature 1/T are depicted. The resistivity values were obtained from the resistors in the equivalent circuits depicted in the insets of Fig. 11a and b. In the case of the HTO sample the open blue symbols represent resistivity values from impedance measurements under Ar atmosphere, which are almost identical to the ones from measurements under air (blue solid symbols). This implies that the ionic oxygen vacancy conduction in HTO may be independent from the ambient conditions.
 | ||
Fig. 12 Resistivity vs. 1/T for the bulk contributions of Gd2Ti2O7 (red squares) and Ho2Ti2O7 (blue squares) ceramics obtained from the equivalent circuit fits. The respective charge transport activation energies were obtained from linear fits (black solid lines) to the Arrhenius plots (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ρ vs. 1/T) and the respective values are given. ρ vs. 1/T) and the respective values are given. | ||
The curve for the GTO ceramic pellet (red solid symbols) shows a deviation from the linear behaviour above ≈500 °C (773 K), which may again be associated with the unusual changes in the GTO lattice parameter at high T (Section 3.3).
The activation energy EA for the GTO ceramic was determined from the respective Arrhenius curves (lnρ vs. 1/T, not shown here) in the linear regime below ≈500 K (773 °C) only, whereas the entire T-range was considered for HTO. EA was found to be 0.79 eV and 0.88 eV for GTO and HTO respectively, which is in a typical range for oxygen ion conduction.49–54 Both EA values are rather similar, which suggests a similar charge transport mechanism. EA was determined by assuming a linear Arrhenius behaviour of ρ = ρ0exp(EA/kBT). However, alternative models of ρ/T = ρ0exp(EA/kBT) have also been proposed in the literature,55,56 in which case the activation energies would be slightly increased to 0.83 eV and 0.92 eV for GTO and HTO respectively. Within the second more advanced model the pre-exponential factors ρ0 have been claimed to exhibit a reciprocal relationship with the concentration of mobile species. From our fits we find ρ0 to be 1.3 10−1 Ω cm K−1 and 8.3 10−5 Ω cm K−1 for GTO and HTO respectively. The concomitant increased concentration of mobile species in HTO is in agreement with the lower nominal resistivity, which may be interpreted in terms of increased oxygen vacancy conduction in agreement with the increased oxygen vacancy disorder in HTO detected by Raman spectroscopy (Section 3.4). Since our ionic conductivity measurements were performed on GTO and HTO pellets sintered at 1400 °C, A and B anti-site defects may well be excluded in both compositions (see Table 1) and the increased oxygen vacancy conduction in HTO may imply a certain decoupling of anti-site and anion Frenkel defects.
Fig. 13 shows the capacitance values obtained from the equivalent circuit fits, plotted as dielectric permittivity ε′ vs. temperature. The ε′ values obtained from the bulk semicircles in GTO and HTO are both in a range that is typical for bulk values.46 The permittivity in the GTO is perceptibly increased as compared to the HTO, despite the equivalent crystal structure and similar cationic composition. The reasons for this increase are unclear, but we suspect that it may be correlated with the anomalous increase of the GTO unit cell (Section 3.3) and the significant changes in the Raman mode intensities (Section 3.4), both with increasing the heat treatment temperature. Although the HTO phase may be more relevant for applications due to an increased ionic conductivity, the GTO sample exhibits interesting unit cell volume changes accompanied by unusual Raman mode intensities, and both GTO and HTO species may deserve further research work in the future.
 | ||
| Fig. 13 Dielectric permittivity vs. temperature data using the capacitance values for the bulk contributions of Gd2Ti2O7 (red squares) and Ho2Ti2O7 (blue squares) ceramics obtained from the equivalent circuit fits. The temperature independent intrinsic bulk dielectric permittivity is indicated by the black lines and the respective values are given. | ||
Both, the resistivity ρ and dielectric permittivity ε′ values presented in Fig. 12 and 13 were reproducible in control samples of GTO and HTO composition within an experimental error of ≈10%, which may arise from the uncertainty in measuring the pellet dimensions to calculate the specific parameters ρ and ε′.
4. Conclusions
We have successfully synthesized the two representatives RE = Gd, Ho from the RE2Ti2O7 pyrochlore family using a microwave assisted synthesis route. The GTO and HTO phases obtained show small particle sizes with a high sintering activity. RE–Ti cationic anti-site defects were found to decrease in concentration with increasing temperature starting from 1100 °C, and completely disappear at 1400 °C. GTO shows a predominantly antiferromagnetic structure, whereas HTO exhibits magnetic saturation and a ferromagnetic component. Impedance spectroscopy data revealed enhanced ionic oxygen vacancy conduction in the HTO sample, which may be associated with oxygen vacancy disorder revealed by Raman spectroscopy that is uncoupled from the anti-site defects.Acknowledgements
We thank the Spanish Ministerio de Ciencia e Innovación and the Comunidad de Madrid for funding the projects MAT2013-64452-C4-1-R and S2013MIT-2753 respectively. Financial support from Universidad CEU San Pablo is acknowledged. We thank the Spanish Ministerio de Ciencia e Innovación (MICINN) and Consejo Superior de Investigaciones Científicas (CSIC) for financial support and for provision of synchrotron radiation facilities. We would like to thank the beamline staff (German Castro y Alvaro Muñoz) for assistance in using beamline BM25-SpLine. R. S. wishes to acknowledge a Ramon y Cajal fellowship from the MICINN/MINECO in Spain. Thanks to Jacobo Santamaría, Carlos León and Alberto Rivera-Calzada for allowing use and help with the Novocontrol impedance spectroscopy facilities at the GFMC.References
- J. E. Greedan, in Functional Oxides, ed. D. W. Bruce, D. O'Hare and R. I. Walton, Wiley, 2011, ch. 2, pp. 41–117 Search PubMed.
- M. Mirsaneh, B. E. Hayden, E. Furman, S. Perini, M. T. Lanagan and I. M. Reaney, Appl. Phys. Lett., 2012, 100, 082901 CrossRef PubMed.
- P. Holtappels, F. W. Poulsen and M. Mogensen, Solid State Ionics, 2000, 135, 675–679 CrossRef CAS.
- M. A. Subramanian, G. Aravamudan and G. V. Subba Rao, Prog. Solid State Chem., 1983, 15, 55–143 CrossRef CAS.
- D. J. P. Morris, D. A. Tennant, S. A. Grigera, B. Klemke, C. Castelnovo, R. Moessner, C. Czternasty, M. Meissner, K. C. Rule, J.-U. Hoffmann, K. Kiefer, S. Gerischer, D. Slobinsky and R. S. Perry, Science, 2009, 326, 411–414 CrossRef CAS PubMed.
- X. W. Dong, K. F. Wang, S. J. Luo, J. G. Wan and J. M. Liu, J. Appl. Phys., 2009, 106, 4 Search PubMed.
- Z. Hiroi and M. Hanawa, J. Phys. Chem. Solids, 2002, 63, 1021–1026 CrossRef CAS.
- S. X. Wang, L. M. Wang, R. C. Ewing and K. V. Govindan Kutty, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 169, 135–140 CrossRef CAS.
- D. P. Reid, M. C. Stennett and N. C. Hyatt, J. Solid State Chem., 2012, 191, 2–9 CrossRef CAS PubMed.
- C. R. Stanek, L. Minervini and R. W. Grimes, J. Am. Ceram. Soc., 2002, 85, 2792–2798 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Nachimuthu, S. Thevuthasan, M. H. Engelhard, W. J. Weber, D. K. Shuh, N. M. Hamdan, B. S. Mun, E. M. Adams, D. E. McCready, V. Shutthanandan, D. W. Lindle, G. Balakrishnan, D. M. Paul, E. M. Gullikson, R. C. C. Perera, J. Lian, L. M. Wang and R. C. Ewing, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 100101 CrossRef.
- B. J. Wuensch, K. W. Eberman, C. Heremans, E. M. Ku, P. Onnerud, E. M. E. Yeo, S. M. Haile, J. K. Stalick and J. D. Jorgensen, Solid State Ionics, 2000, 129, 111 CrossRef CAS.
- J. Chen, J. Lian, L. M. Wang, R. C. Ewing, R. G. Wang and W. Pan, Phys. Rev. Lett., 2002, 88, 105901 CrossRef CAS.
- P. K. Moon and H. L. Tuller, MRS Symposia Proceedings No. 135: Solid State Ionics, Pittsburgh, 1989, p. 149 Search PubMed.
- A. Rivera-Calzada, M. R. Diaz-Guillen, O. J. Dura, G. Sanchez-Santolino, T. J. Pennycook, R. Schmidt, F. Y. Bruno, J. Garcia-Barriocanal, Z. Sefrioui, N. M. Nemes, M. Garcia-Hernandez, M. Varela, C. Leon, S. T. Pantelides, S. J. Pennycook and J. Santamaria, Adv. Mater., 2011, 23, 5268–5274 CrossRef CAS PubMed.
- K. J. Moreno, A. F. Fuentes, U. Amador, J. Santamarıa and C. Leon, J. Non-Cryst. Solids, 2007, 353, 3947–3955 CrossRef CAS PubMed.
- K. J. Moreno, A. F. Fuentes, M. Maczka, J. Hanuza, U. Amador, J. Santamaría and C. León, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 184303 CrossRef.
- A. F. Fuentes, K. Boulahya, M. Maczka, J. Hanuza and U. Amador, Solid State Sci., 2005, 7, 343–353 CrossRef CAS PubMed.
- A. Garbout, I. Ben Taazayet-Belgacem and M. Férid, J. Alloys Compd., 2013, 573, 43–52 CrossRef CAS PubMed.
- M. Roy, I. Bala and S. K. Barbar, J. Therm. Anal. Calorim., 2012, 110, 559–565 CrossRef CAS.
- R. S. Pavlov, J. B. C. Castelló, V. B. Marzá and J. M. Hohembergerger, J. Am. Ceram. Soc., 2002, 85, 1197–1202 CrossRef CAS PubMed.
- A. V. Shlyakhtina, O. K. Karyagina and L. G. Shcherbakova, Inorg. Mater., 2004, 40, 59–65 CrossRef CAS.
- L. Zhang, H. Zhong, W. Zhang, L. Lu, X. Yang and X. Wang, J. Alloys Compd., 2008, 463, 466–470 CrossRef CAS PubMed.
- M. L. Hand, M. C. Stennett and N. C. Hyatt, J. Eur. Ceram. Soc., 2012, 32, 3211–3219 CrossRef CAS PubMed.
- X. Lu, Y. Ding, H. Dan, S. Yuan, X. Mao, L. Fan and Y. Wu, Ceram. Int., 2014, 40, 13191–13194 CrossRef CAS PubMed.
- J. Zhao and W. Yan, in Modern Inorganic Synthetic Chemistry, Elsevier, Amsterdam, 2011, pp. 173–195, DOI:10.1016/b978-0-444-53599-3.10008-3.
- M. Gupta and E. W. W. Leong, Microwaves and Metals, Wiley, 2008 Search PubMed.
- J. Prado-Gonjal, A. M. Arévalo-López and E. Morán, Mater. Res. Bull., 2011, 46, 222–230 CrossRef CAS PubMed.
- J. Prado-Gonjal, R. Schmidt, J.-J. Romero, D. Ávila, U. Amador and E. Morán, Inorg. Chem., 2013, 52, 313–320 CrossRef CAS PubMed.
- J. Prado-Gonjal, R. Schmidt and E. Morán, Inorganics, 2015, 3, 101 CrossRef CAS PubMed.
- R. Schmidt, J. Prado-Gonjal and E. Morán, in CRC Concise Encyclopedia of Nanotechnology, ed. B. Kharisov, O. Kharissova and U. Ortiz-Mendez, CRC Press Taylor & Francis Group, Boca Raton (USA), 2015, ch. 46 Search PubMed.
- J. Prado-Gonjal, R. Schmidt and E. Morán, in Perovskite: Crystallography, Chemistry and Catalytic Performance, ed. J. Zhang and H. Li, Nova Science Pub Incorporated, 2012, pp. 117–140 Search PubMed.
- J. Rodriguez-Carvajal, Satellite Meeting on Powder diffraction of the XV Congress of the IUCr, 1990 Search PubMed.
- E. Barsukov and J. Macdonald, Impedance Spectroscopy: Theory, Experiment and Applications, John Wiley & Sons Inc., Hoboken, 2005 Search PubMed.
- P. K. Moon and H. L. Tuller, MRS Online Proc. Libr., 1988, 135, 149, DOI:10.1557/proc-135-149.
- D. V. West, T. M. McQueen, Q. Huang and R. J. Cava, J. Solid State Chem., 2008, 181, 1753–1758 CrossRef CAS PubMed.
- G. Sala, M. J. Gutmann, D. Prabhakaran, D. Pomaranski, C. Mitchelitis, J. B. Kycia, D. G. Porter, C. Castelnovo and J. P. Goff, Nat. Mater., 2014, 13, 488–493 CrossRef CAS PubMed.
- M. T. Vandenborre, E. Husson, J. P. Chatry and D. Michel, J. Raman Spectrosc., 1983, 14, 63–71 CrossRef CAS PubMed.
- F. W. Poulsen, M. Glerup and P. Holtappels, Solid State Ionics, 2000, 135, 595–602 CrossRef CAS.
- B. P. Mandal, A. Banerji, V. Sathe, S. K. Deb and A. K. Tyagi, J. Solid State Chem., 2007, 180, 2643–2648 CrossRef CAS PubMed.
- M. Glerup, O. F. Nielsen and F. W. Poulsen, J. Solid State Chem., 2001, 160, 25–32 CrossRef CAS.
- K. Matsuhira, Y. Hinatsu, K. Tenya and T. Sakakibara, J. Phys.: Condens. Matter, 2000, 12, L649 CrossRef CAS.
- T. Fennell, P. P. Deen, A. R. Wildes, K. Schmalzl, D. Prabhakaran, A. T. Boothroyd, R. J. Aldus, D. F. McMorrow and S. T. Bramwell, Science, 2009, 326, 415–417 CrossRef CAS PubMed.
- A. P. Sazonov, A. Gukasov and I. Mirebeau, J. Phys.: Condens. Matter, 2011, 23, 164221 CrossRef CAS PubMed.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132 CrossRef CAS PubMed.
- E. J. Abram, D. C. Sinclair and A. R. West, J. Electroceram., 2003, 10, 165 CrossRef CAS.
- R. Schmidt, W. Eerenstein, T. Winiecki, F. D. Morrison and P. A. Midgley, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 245111 CrossRef.
- J. Prado-Gonjal, R. Schmidt, J. Espíndola-Canuto, P. Ramos-Alvarez and E. Morán, J. Power Sources, 2012, 209, 163–171 CrossRef CAS PubMed.
- M. A. Frechero, O. J. Durá, M. R. Díaz-Guillén, K. J. Moreno, J. A. Díaz-Guillén, J. García-Barriocanal, A. Rivera-Calzada, A. F. Fuentes and C. León, J. Non-Cryst. Solids, 2015, 407, 349–354 CrossRef CAS PubMed.
- L. G. Shcherbakova, J. C. C. Abrantes, D. A. Belov, E. A. Nesterova, O. K. Karyagina and A. V. Shlyakhtina, Solid State Ionics, 2014, 261, 131–140 CrossRef CAS PubMed.
- J. Prado-Gonjal, R. Heuguet, D. Muñoz-Gil, A. Rivera-Calzada, S. Marinel, E. Morán and R. Schmidt, Int. J. Hydrogen Energy, 2015 DOI:10.1016/j.ijhydene.2015.07.161.
- M. R. Díaz-Guillén, M. A. Frechero, J. A. Díaz-Guillén, A. F. Fuentes and C. León, J. Electroceram., 2015, 34, 15–19 CrossRef.
- M. Stojmenović, M. Žunić, J. Gulicovski, D. Bajuk-Bogdanović, I. Holclajtner-Antunović, V. Dodevski and S. Mentus, J. Mater. Sci., 2015, 50, 3781–3794 CrossRef.
- B. P. Mandal, S. K. Deshpande and A. K. Tyagi, J. Mater. Res., 2008, 23, 911–916 CrossRef CAS.
- K. J. Moreno, G. Mendoza-Suárez, A. F. Fuentes, J. García-Barriocanal, C. León and J. Santamaria, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 132301 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |