
Controllable synthesis of porous iron–nitrogen–carbon nanofibers with enhanced oxygen reduction electrocatalysis in acidic medium†
X. X. Yan,
L. Gan,
F. Fang,
K. X. Liu,
J. Luo* and
J. Zhu*
National Center for Electron Microscopy in Beijing, Key Laboratory of Advanced Materials (MOE), The State Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China. E-mail: luojunkink@126.com; jzhu@mail.tsinghua.edu.cn
First published on 26th May 2015
Abstract
We synthesized porous Fe–N–C nanofibers as non-precious metal catalysts to investigate the impact of surface area on electrocatalysis performance. The surface area was modified by adjusting the proportion of the added silicon nanoparticles, and achieved a 20 times enhancement of the electrocatalysis performance at an optimized proportion.
As one of the most significant problems nowadays, the energy crisis has aroused interest in a wide variety of research on new energy conversion devices, such as fuel cells (FCs).1–3 However, the commercialization and widespread application of FCs are seriously restricted by the high usage of expensive platinum-based catalysts, which have been applied to improve the sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode.4,5 Various types of non-precious metal (NPM) catalysts with low cost have been discovered and developed to replace those Pt-based catalysts for many decades.6–9 Among them, the most promising alternative to Pt-based catalysts are those which contain active sites of transition metal coordinated with nitrogen atoms (TM-Nx) on carbon supports, especially when the medium is acidic, in order to match the conditions of a proton exchange membrane fuel cell (PEMFC).10–14 Since the first report of such an ORR-activated structure in 1964,6 such NPM catalysts have been greatly improved during the last decade.10–12 This research has pointed out that the surface area and active sites play signification roles in electrocatalysis performance. However, in the reported impregnation method,10–12 there is still a trade-off remaining between the surface area and the active sites. For one thing, the process of absorbing active sites will cause an obvious drop in surface area.11,15 For another, the treatment of increasing the surface area will damage the active sites.16 Ammonia treatment has been developed to supply nitrogen atoms for remaining active sites in addition to increasing the surface area.11,17 However, ammonia is dangerous and not very environmentally friendly. Hence, it seems that is difficult to obtain both a high surface area and an abundance of active sites at the same time. In this work, we have synthesized a new type of porous iron–nitrogen–carbon (Fe–N–C) nanofiber as a NPM catalyst by removing silicon nanoparticles (Si NPs) in carbon nanofibers, which were employed to create a porous structure, thereby acquiring a high surface area and plenty of active sites. We applied them in an acidic medium, and revealed that the electrocatalysis performance of these catalysts could be enhanced, after etching Si NPs, and controlled, by modification to create an additional porous structure. With the use of transmission electron microscopy (TEM) observations, we further found that, with the increment in the weight proportion of Si NPs, the electrochemical performances of various porous catalysts were firstly enhanced and then dropped due to the aggregation of Si NPs.
As depicted in Fig. 1a, porous Fe–N–C nanofibers were synthesized by first electrospinning polyacrylonitrile (PAN) nanofibers containing ferric nitrate and Si NPs,18,19 then heating the as-electrospun nanofibers to get carbon nanofibers,20,21 and finally etching the Si NPs in an alkaline solution (typically 3 M KOH) to create the expected porous structure (see experimental details in the ESI†). Our method is compatible with the carbon fiber industry, as the major precursor is PAN, which is an easily acquired industrial material.22 The weight ratio of the Si NPs and PAN (x = Si/PAN) was set from 0 to 2.5, with increments of 0.5, and therefore the final samples were named as PFeNC05, PFeNC10, PFeNC15, PFeNC20 and PFeNC25, respectively. The sample named FeNC was the one with no added Si NPs and no etching process.
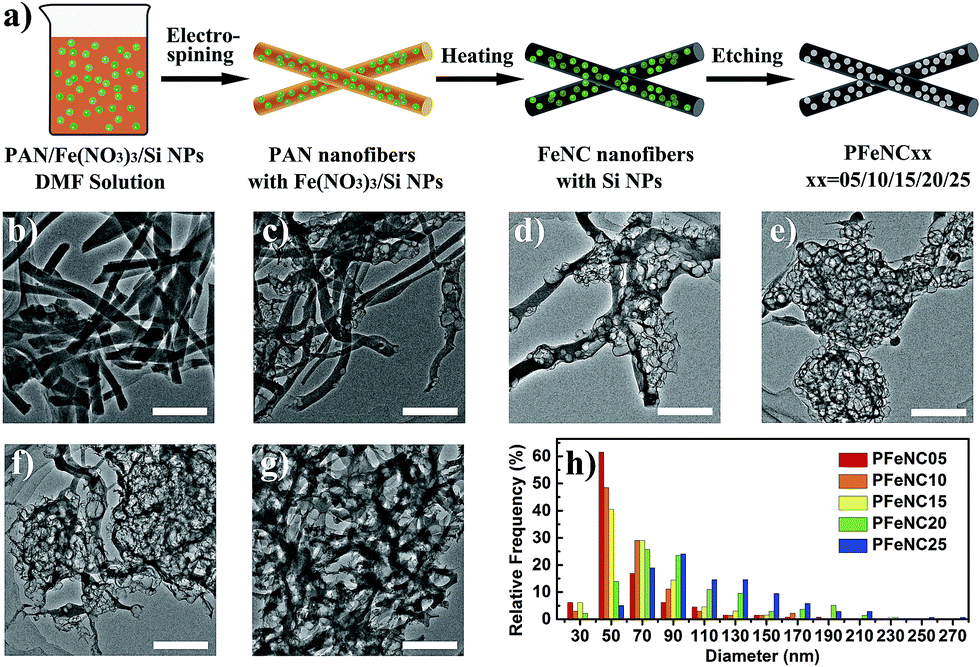 | ||
| Fig. 1 (a) Schematic diagram of the synthesis of porous Fe–N–C nanofibers by electrospinning, heating and etching of Si NPs (green balls). (b–g) The typical TEM images of FeNC, PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25 samples, respectively. The scale bars are all 500 nm. (g) Histograms between the relative frequency and pore diameters of PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25, respectively. | ||
Fig. 1b depicts a TEM image of the FeNC sample without a porous structure. FeNC samples were mainly nanofibers about 200 nm in diameter. Fig. 1c–g are the TEM images of samples with various Si/PAN ratios: PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25. These indicate that the samples also kept the nanofiber morphology and pores appeared in the nanofibers. Because of the relatively low proportion of added Si NPs, the pores of PFeNC05 were distributed sparsely, as shown in Fig. 1c, matching with the TEM image before etching shown in Fig. S1a of the ESI.† With the increment in the Si/PAN ratio, the density of the distributed pores increased. The samples with a high Si/PAN ratio contained an even denser distribution of pores in the nanofibers, as depicted in Fig. 1e–g. After detailed calibration of the pore diameters, we compared different histograms between the relative frequency and the pore diameter of PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25, as shown in Fig. 1h. Pores with a diameter of about 50 nm were most frequent for the PFeNC05 sample. Thus, the Si NPs were dispersed separately in the nanofibers of PFeNC05. However, some values of pore diameter for the PFeNC05 sample were higher than 50 nm, indicating that some of the Si NPs aggregated in the nanofibers. The mean pore diameters (〈DP〉) of PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25 were 62, 66, 68, 97 and 112 nm, respectively. These data reveal that the probability of aggregation of the Si NPs in the nanofibers became higher after adding more Si NPs. This could be related to space limitation in the cross-section of the nanofiber structure. Fig. S1b in the ESI† shows a TEM image of PFeNC15 samples before etching, indicating that the aggregation of Si NPs could cause the distortion of the nanofibers. Fig. S2–S4 in the ESI† provide more TEM images of PFeNC15, PFeNC20 and PFeNC25 to complement those in Fig. 1e–g, revealing that the morphology of the nanofibers was maintained with many large pores. The maximum value of the pore diameter reached 270 nm, which was equal to the size of an aggregate of at least 38 Si NPs (Fig. S5 in ESI†). This high degree of aggregation may be explained by the small particles coagulating in the fluid precursor solution during the electrospinning process.23–25
After heat treatment, PAN can convert to a conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C conjugation structure with doped nitrogen, which can be coordinated with iron to form Fe-Nx active sites, as suggested by the results of thermal gravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS), shown in Fig. S6 of the ESI.†26–29 Fig. 2 shows the electrochemical measurement of porous Fe–N–C nanofiber catalysts in an acidic medium (0.1 M HClO4 solution). In such a medium, the ORR happened following the reaction equation of O2 + 4H+ + 4e− = 2H2O.4 Fig. 2a shows that, after etching of the Si NPs, the current density of PFeNC05 was increased from 0.037 mA cm−2 (0 h) to 0.237 mA cm−2 (72 h) at 0.6 V versus a reversible hydrogen electrode (RHE), and enhanced by 6.4 times. With a longer etching time, the current density was higher, indicating that a more porous structure could increase the electrocatalysis performance. In our experimental conditions, the best etching time was about 72 h to remove the Si NPs entirely, and further etching couldn’t increase the performance. TEM observation of the etched samples in Fig. 1c–g also confirmed that no Si NPs remained in the nanofibers. In order to exclude the etching effect on the carbon nanofiber matrix, we utilized the same method to treat the FeNC sample. Polarization curves of the FeNC without and with etching treatment were almost the same, as displayed in Fig. 2b. Therefore, the enhancement of the current density, as shown in Fig. 2a, was primarily caused by the additional surface area of the porous structure. Furthermore, scanning electron microscopy (SEM) images and energy dispersive X-ray (EDX) spectra of the PFeNC05 samples before and after creating pores are shown in Fig. 2c–f. From the SEM images in Fig. 2c and e, it is observed that the samples kept their nanofiber morphology, as was also shown by the TEM observations, and the Si NPs were totally removed after etching for 72 h, as shown by the EDX results in Fig. 2d and f. Furthermore, the element content determined by XPS analysis, shown in Table S1 of the ESI,† denotes that, after etching, the Si atoms were almost completely removed, in accordance with the EDX results, and the number of iron atoms was decreased. The etching process could remove the inactive compound containing iron, which has no ORR activity.12 This explanation could be confirmed by the results in Fig. 2b, which show that the etching treatment could not affect the ORR activity of the FeNC samples, as determined by measurements before and after the etching treatment.
C conjugation structure with doped nitrogen, which can be coordinated with iron to form Fe-Nx active sites, as suggested by the results of thermal gravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS), shown in Fig. S6 of the ESI.†26–29 Fig. 2 shows the electrochemical measurement of porous Fe–N–C nanofiber catalysts in an acidic medium (0.1 M HClO4 solution). In such a medium, the ORR happened following the reaction equation of O2 + 4H+ + 4e− = 2H2O.4 Fig. 2a shows that, after etching of the Si NPs, the current density of PFeNC05 was increased from 0.037 mA cm−2 (0 h) to 0.237 mA cm−2 (72 h) at 0.6 V versus a reversible hydrogen electrode (RHE), and enhanced by 6.4 times. With a longer etching time, the current density was higher, indicating that a more porous structure could increase the electrocatalysis performance. In our experimental conditions, the best etching time was about 72 h to remove the Si NPs entirely, and further etching couldn’t increase the performance. TEM observation of the etched samples in Fig. 1c–g also confirmed that no Si NPs remained in the nanofibers. In order to exclude the etching effect on the carbon nanofiber matrix, we utilized the same method to treat the FeNC sample. Polarization curves of the FeNC without and with etching treatment were almost the same, as displayed in Fig. 2b. Therefore, the enhancement of the current density, as shown in Fig. 2a, was primarily caused by the additional surface area of the porous structure. Furthermore, scanning electron microscopy (SEM) images and energy dispersive X-ray (EDX) spectra of the PFeNC05 samples before and after creating pores are shown in Fig. 2c–f. From the SEM images in Fig. 2c and e, it is observed that the samples kept their nanofiber morphology, as was also shown by the TEM observations, and the Si NPs were totally removed after etching for 72 h, as shown by the EDX results in Fig. 2d and f. Furthermore, the element content determined by XPS analysis, shown in Table S1 of the ESI,† denotes that, after etching, the Si atoms were almost completely removed, in accordance with the EDX results, and the number of iron atoms was decreased. The etching process could remove the inactive compound containing iron, which has no ORR activity.12 This explanation could be confirmed by the results in Fig. 2b, which show that the etching treatment could not affect the ORR activity of the FeNC samples, as determined by measurements before and after the etching treatment.
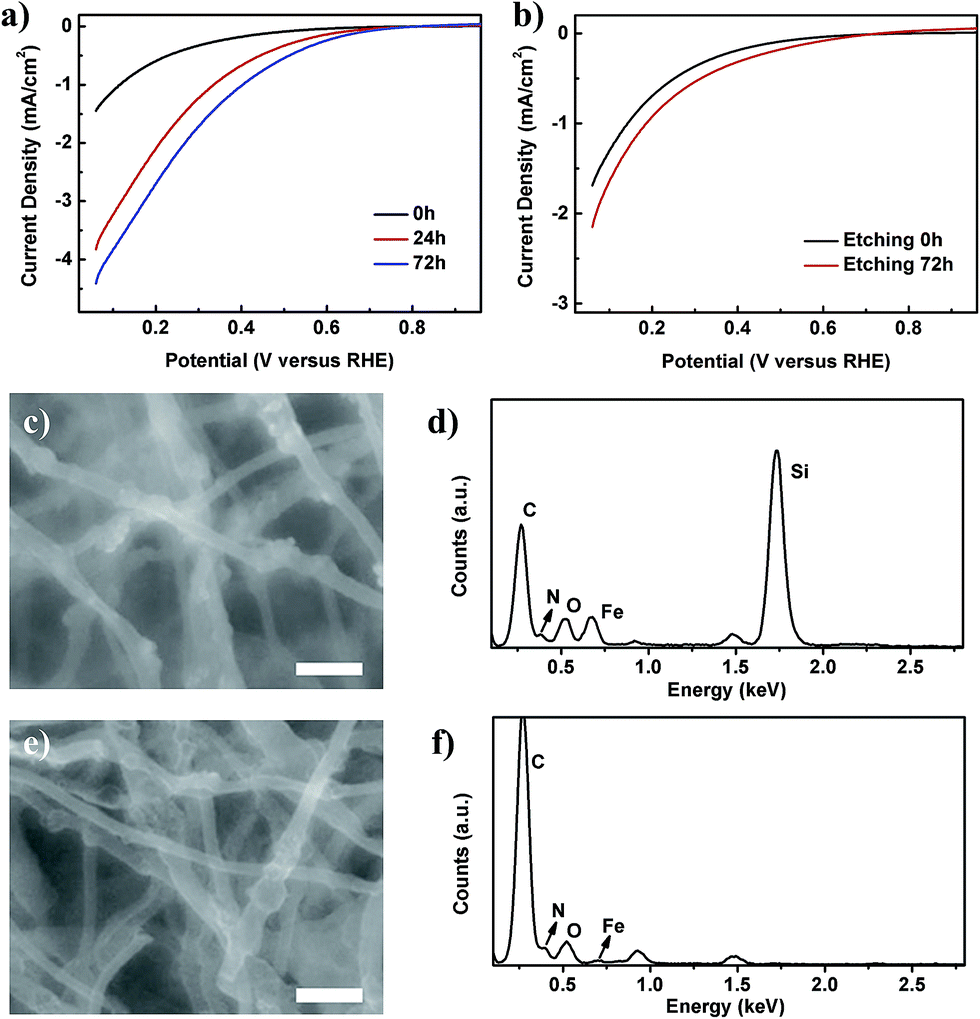 | ||
| Fig. 2 Results of the etching of Si NPs in the nanofibers. (a) The linear sweep voltammetry (LSV) curves of the PFeNC05 samples with different etching times. (b) The LSV curves of the FeNC samples without and with the etching process. (c and d) SEM image of nanofibers without etching, and the corresponding EDX spectrum, respectively. (e and f) SEM image of nanofibers after etching for 72 h, and the corresponding EDX spectrum, respectively. The scale bars in (c) and (e) are 500 nm. | ||
Fig. 3a shows the electrochemical performances of all the porous catalysts: FeNC, PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25. Comparing with other porous Fe–N–C nanofiber catalysts, the FeNC sample had the lowest performance. For further comparison of various polarization curves, we plot the absolute current density at 0.6 V versus RHE against Si/PAN in Fig. 3b. With the increment of the Si/PAN ratio, the absolute current densities of the ORR were firstly enhanced, reached the maximum value of 0.80 mA cm−2 at Si/PAN = 1.5, and then dropped. Comparatively, the maximum current density of the porous catalysts was 20 times that of the FeNC catalysts (0.39 mA cm−2). As reported, the performance is related to the surface area.11,13,27 The current density was expected to keep increasing after adding more Si NPs. However, the experimental results indicate that there was an optimal value of Si/PAN. The TEM observations indicate that aggregation may limit the enhancement of the porosity of the structure. It can be concluded that the total specific surface area (SSA) of the porous catalysts (S) consists of the SSA of the nanofibers (SNF) and the additional SSA of the porous structure (SP):
| S = SNF + SP. | (1) |
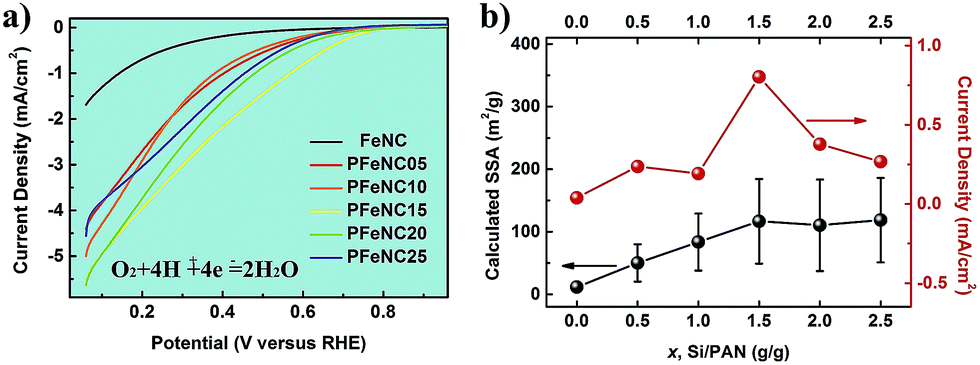 | ||
| Fig. 3 (a) The LSV curves of FeNC, PFeNC05, PFeNC10, PFeNC15, PFeNC20, and PFeNC25, respectively. (b) The dependence of the current density at 0.6 V versus RHE and the calculated SSA from the mean pore diameter in the nanofibers on the weight ratio of the Si NPs and PAN (Si/PAN). | ||
SNF was proved to be the same in various samples, as the etching process didn’t affect the surface area of the carbon nanofibers as discussed above, and could be calculated in a cylinder model:
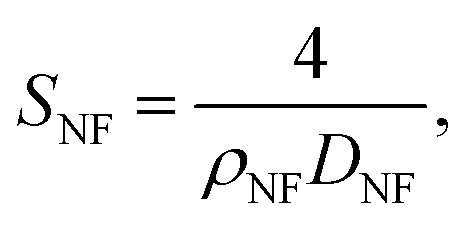 | (2) |
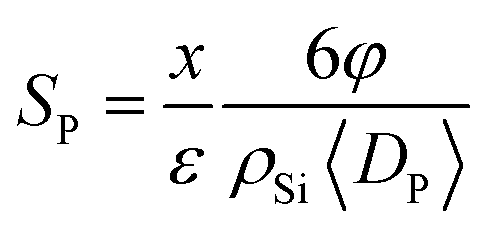 | (3) |
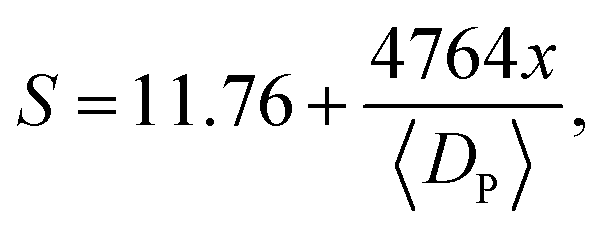 | (4) |
Furthermore, we normalized the activities (current densities) of all samples by calculated SSA. Fig. S8 in the ESI† shows that all normalized values were the same as each other, indicating that the ORR performance difference was only caused by the SSA. It could be inferred that only the surface areas of the various samples were different, and the active sites of Fe-Nx were almost unchanged after the etching treatment, in our experiments. Hence, the new surfaces created by removing the Si NPs were also decorated with the active sites. The catalytic activity of the porous catalysts had a positive relationship with the surface area of the additional pores, whose pore sizes were 50 nm and larger. From this deduction, some researchers hold the view that the catalytic activity was proportional to the microporous surface area of the catalysts, whose pore size was less than 2 nm.13,27 Then, we suggested that, not only the micropores but also the larger pores can provide the surface area with active sites and improve the mass transport of substance and production. More importantly, there is an optimization of such additional surface area from the etching of the NPs. In our experiments, the optimized weight ratio of Si NPs and PAN was 1.5, which enhanced the calculated specific surface area and current density 10-fold and 20-fold, respectively.
Conclusions
In summary, we synthesized porous Fe–N–C nanofibers as non-precious metal catalysts by a facile and low-cost method. The porous structure can be created by etching the added silicon nanoparticles in the nanofibers, without affecting the carbon nanofibers. The current density of the etched nanofibers increased with longer etching time. Furthermore, the surface area was controlled by modifying the proportion of the added Si nanoparticles, which realized a 20 times enhancement of the electrocatalysis performance at an optimized weight ratio of Si NPs and PAN (Si/PAN = 1.5). These results indicated that the porous structure with pore sizes of about 50 nm could improve the surface area of the nanofibers and enhance the electrocatalysis performance. Nevertheless, this method can also be applied to increase both the surface area and active sites for materials in other fields.Acknowledgements
This work was financially supported by the National 973 Project of China (2015CB654902), the Chinese National Natural Science Foundation (11374174, 51390471 and 51102145), and the National Program for Thousand Young Talents of China. This work made use of the resources of the National Center for Electron Microscopy in Beijing.Notes and references
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed
.
- N. Demirdöven and J. Deutch, Science, 2004, 305, 974–976 CrossRef PubMed
- U. Eberle, B. Müller and R. von Helmolt, Energy Environ. Sci., 2012, 5, 8780–8798 Search PubMed
- L. Carrette, K. A. Friedrich and U. Stimming, ChemPhysChem, 2000, 1, 162–193 CrossRef CAS
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS PubMed
- N. A. Vante and H. Tributsch, Nature, 1986, 323, 431–432 CrossRef CAS PubMed
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed
- M. Risch, K. A. Stoerzinger, S. Maruyama, W. T. Hong, I. Takeuchi and Y. Shao-Horn, J. Am. Chem. Soc., 2014, 136, 5229–5232 CrossRef CAS PubMed
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed
- M. Lefèvre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed
- F. Jaouen, M. Lefèvre, J. P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed
- Z. Liu, G. Zhang, Z. Lu, X. Jin, Z. Chang and X. Sun, Nano Res., 2013, 6, 293–301 CrossRef CAS PubMed
- G. S. Chai, I. S. Shin and J. S. Yu, Adv. Mater., 2004, 16, 2057–2061 CrossRef CAS PubMed
- G. Faubert, R. Côté, J. P. Dodelet, M. Lefèvre and P. Bertrand, Electrochim. Acta, 1999, 44, 2589–2603 CrossRef CAS
- J. Yin, Y. J. Qiu and J. Yu, Electrochem. Commun., 2013, 30, 1–4 CrossRef CAS PubMed
- M. Inagaki, Y. Yang and F. Y. Kang, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed
- X. X. Yan, L. Gan, Y.-C. Lin, L. Bai, T. Wang, X. Q. Wang, J. Luo and J. Zhu, Small, 2014, 10, 4072–4079 CAS
- W. X. Zhang, J. Liu and G. Wu, Carbon, 2003, 41, 2805–2812 CrossRef CAS
- J. B. Donnet and R. C. Bansal, Carbon Fibers, Marcel Dekker, New York, 1984 Search PubMed
- E. Frank, L. M. Steudle, D. Ingildeev, J. M. Spörl and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2014, 53, 5262–5298 CrossRef CAS PubMed
- D. A. Weitz and M. Oliveria, Phys. Rev. Lett., 1984, 52, 1433–1436 CrossRef CAS
- Y. M. Xuan, Q. Li and W. F. Hu, AIChE J., 2003, 49, 1038–1043 CrossRef CAS PubMed
- A. Serov, K. Artyushkova and P. Atanassov, Adv. Energy Mater., 2014, 4, 1301735 Search PubMed
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5531–5543 CrossRef CAS
- F. Jaouen, J. Herranz, M. Lefèvre, J. P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CAS
- L. F. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. H. Tang, H. Gong, Z. X. Shen, L. Y. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS
- S. Gupta, D. Tryk, I. Bae, W. Aldred and E. Yeager, J. Appl. Electrochem., 1989, 19, 19–27 CrossRef CAS
- X. X. Yan, K. X. Liu, X. Q. Wang, T. Wang, J. Luo and J. Zhu, Nanotechnology, 2015, 26, 165401 CrossRef PubMed
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 82nd edn, 2002 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and Fig. S1 and S2. See DOI: 10.1039/c5ra07741a |
| This journal is © The Royal Society of Chemistry 2015 |