
Beneficiary effect of nanosizing ferric pyrophosphate as food fortificant in iron deficiency anemia: evaluation of bioavailability, toxicity and plasma biomarker†
Abstract
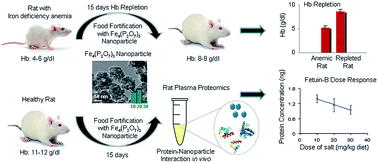
Iron deficiency anemia is a global health issue affecting a significant population worldwide. Fortification of food with iron compounds is a widely used strategy to prevent iron deficiency anemia. To overcome sensory effects, water insoluble and white colored compounds, like ferric pyrophosphate, are used to fortify infant cereals. Ferric pyrophosphate is poorly bioavailable due to its low solubility even in an acid medium. Nanosized iron salts find potential applications in food fortification, since solubility increases with decrease in the particle size. However, limited knowledge exists about the effect of nanoparticles in a biological system. The present study addresses the efficacy of synthesized ferric pyrophosphate in its nano form (10–30 nm) as a potential food fortificant in iron deficiency anemia and measures its toxicity in a rat model. Additionally, the effect of the nanoparticle in vivo has been explored using a mass-spectrometry-based plasma proteomics approach. The relative bioavailability of ferric pyrophosphate nanoparticle, calculated using hemoglobin regeneration efficiency was found to be 103.02% with respect to the reference salt, ferrous sulphate. Histopathological examinations of different organs did not show any significant toxicity attributable to nanoparticle ferric pyrophosphate. However, plasma proteomics analysis showed a decreasing trend in Fetuin-B concentration with increasing dose levels of nanoparticle ferric pyrophosphate. In conclusion, the nanoparticle ferric pyrophosphate could be a promising food fortificant in combating iron deficiency anemia, while Fetuin-B, a negative acute phase protein, might be a potential candidate for detecting biological responses to the nanoparticle exposure in vivo.
 Please wait while we load your content...
Please wait while we load your content...

