
Influence of pH on the catalytic performance of CuO–CoOx–CeO2 for CO oxidation
Shupanxiang Chena,
Sufang Zhao†
a,
Zhen Xuc,
Zhigang Liu*a and
Runliang Zhu*b
aSchool of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, China. E-mail: liuzhigang@hnu.edu.cn; Tel: +86-731-8882-3327
bGuangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou, Guangdong 510640, China. E-mail: 33882506@qq.com
cSchool of Materials Science and Engineering, Central South University, Changsha, 410083, China
First published on 29th June 2015
Abstract
Ternary catalysts, namely CuO–CoOx–CeO2, are prepared by coprecipitation with different pH and characterized by techniques such as N2 adsorption/desorption, XRD, TPR, TEM and XPS. The results show that pH plays a crucial role in determining the physicochemical properties such as surface area, particle sizes and valence states of the elements on the surface of the catalysts. It can also be found that suitable pH values may facilitate the formation of Cu+ and the redox recycle of Cu2+ to Cu+ due to the synergistic effect between copper oxide and the support such as cobalt oxide and ceria, which, as a result, is beneficial to achieve CuO–CoOx–CeO2 with high catalytic performance for CO oxidation.
1. Introduction
Cerium oxide and ceria-containing materials have attracted great interest and been used as catalysts for catalytic oxidation such as CO oxidation due to their capacity in oxygen storage and release.1–6 When transition metal oxides such as CuO and CoOx are mixed or/and doped with ceria, their catalytic performance is dramatically enhanced, which is mainly attributed to the synergistic redox properties of the interfacial interaction among CuO, CoOx and CeO2.7–12Taking CuO/CeO2 as example, Wang et al.13 reported that the catalytic activity is governed both by the Cu+/Cu2+ and Ce3+/Ce4+ redox couples. At the same time, Teng14 and Liotta15 have synthesized catalysts including CuO/CoOx and CoOx/CeO2. They found that these catalysts exhibit remarkable catalytic performance for CO oxidation, which is also attributed to the interfacial interaction among CuO, CoOx and CeO2.
Because of the interaction between the active sites and supports, CuO species in both fully oxidized and partially reduced states are significantly affected by the interaction with underlying CeO2 support and the degree of promotion of such reduction could be also affected by modifying the nature of the support.16–19 Evidently, the catalytic properties can be strongly affected by the synthetic methodology.20,21 Herein, preparation methods usually play a crucial role in determining the properties of catalysts.
For example, Woods et al.22 prepared CoOx/CeO2 with high surface area using precipitation and incipient wetness impregnation method. This catalyst had high specific surface area and significantly high Co dispersion (3.4%) over nano-particles of CeO2. Gawade et al.23 prepared ceria with high surface area through a precipitation technique and Co incorporation was achieved using a wet impregnation method. It is found that higher Co loadings are favored higher CO oxidation at the expense of oxygen selectivity. Moreover, the Co phase is identified as Co3O4 and is stable in the reducing environments. Arango-Díaz et al.24 reported that nanocrystalline ceria with particle sizes of 9.5 nm was prepared by a freeze-drying method and subsequently impregnated copper precursors. The resulting CuO/CeO2 exhibited a good catalytic performance for CO oxidation in rich hydrogen despite their low specific surface areas. They attributed this to the wide dispersion of the copper active sites associated with the high amount of Ce4+ species before reaction. Chen et al.25 prepared Co3O4–CeO2 through a coprecipitation method and copper precursors were impregnated on Co3O4–CeO2. They found multiple Cu species including Cu0, Cu+ and Cu2+ species coexist in the spent catalyst. We also investigated the influence of preparation methods on the properties of catalysts.26–28 A chelating method is employed to synthesize CuO–CeO2 catalyst, which is found to have remarkable catalytic performance for CO oxidation in rich hydrogen. Moreover, the influence of pH value is investigated.27 We find that pH values of the solution during the preparation of the catalysts have an important effect on the properties of the catalysts such as particle size, specific surface area. However, have the physicochemical properties of the ternary catalysts been influenced by the pH?
In this study, we prepare CuO–CoOx–CeO2 catalysts by a coprecipitation method and the influence of pH values on the properties of the catalysts has been investigated. Techniques such as N2 adsorption/desorption, XRD, TPR, TEM and XPS are used to explore the nature of the physicochemical properties of the catalysts. As the influence of pH values on the valence states of the metal oxides is focused, this work exhibits an interesting viewpoint to explain the effect of pH values on the physicochemical properties and catalytic performance of the ternary catalysts for CO oxidation.
2. Experimental
2.1 Catalysts preparation
CuO–CoOx–CeO2 is prepared by coprecipitation method, with Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co:Ce atomic ratios equal to 1:5:5. 100 ml NaOH solution (1.20 g NaOH dissolved in 100 ml deionized water) is dropwise added to a mixed solution (0.2403 g Cu(NO3)2·3H2O with corresponding amount of CoCl2·6H2O and Ce(NO3)3·6H2O dissolved in 100 ml deionized water) under vigorous stirring. After stirring for about 30 min, the obtained product is filtered and washed, first with 100 ml deionized water and then 100 ml anhydrous ethanol. Then the washed precipitate is dried at 80 °C for about 3 h before treated at 500 °C for 1 h. The obtained sample is designated as CCC(1.2) in which 1.2 means the mass of NaOH. As described in above process, samples including CCC(1.4), CCC(1.6), CCC(1.8) and CCC(1.9) have been prepared. The pH values of precipitant solution are listed in Table 1. And adopting the similar method, CoOx–CeO2(1.8) and CeO2(1.8) samples are prepared.
:Co:Ce atomic ratios equal to 1:5:5. 100 ml NaOH solution (1.20 g NaOH dissolved in 100 ml deionized water) is dropwise added to a mixed solution (0.2403 g Cu(NO3)2·3H2O with corresponding amount of CoCl2·6H2O and Ce(NO3)3·6H2O dissolved in 100 ml deionized water) under vigorous stirring. After stirring for about 30 min, the obtained product is filtered and washed, first with 100 ml deionized water and then 100 ml anhydrous ethanol. Then the washed precipitate is dried at 80 °C for about 3 h before treated at 500 °C for 1 h. The obtained sample is designated as CCC(1.2) in which 1.2 means the mass of NaOH. As described in above process, samples including CCC(1.4), CCC(1.6), CCC(1.8) and CCC(1.9) have been prepared. The pH values of precipitant solution are listed in Table 1. And adopting the similar method, CoOx–CeO2(1.8) and CeO2(1.8) samples are prepared.
| Samples | pHa | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) | Crystallite sizeb (nm) | ||
|---|---|---|---|---|---|---|---|
| CeO2 | Co3O4 | CuO | |||||
| a The pH values of precipitant solution.b Crystallite sizes of CeO2, Co3O4 and CuO were calculated by using Scherrer equation from CeO2 (111), Co3O4 (311) and CuO (002) reflection, respectively. | |||||||
| CCC(1.2) | 12.1 | 92.7 | 0.26 | 11.4 | 10.4 | 16.0 | — |
| CCC(1.4) | 12.5 | 103.5 | 0.24 | 9.3 | 11.2 | 17.2 | — |
| CCC(1.6) | 12.7 | 131.8 | 0.42 | 12.7 | 9.4 | 18.0 | — |
| CCC(1.8) | 12.9 | 127.6 | 0.60 | 18.8 | 8.9 | 20.7 | — |
| CCC(1.9) | 12.9 | 100.9 | 0.29 | 11.4 | 11.1 | 20.3 | 18.3 |
2.2 Evaluation of catalytic activity
The catalytic activity of the catalysts for CO oxidation is measured in a continuous fixed-bed micro-reactor. 50 mg catalyst is loaded in a quartz tube reactor (i.d. 4 mm) without any pretreatment. The feed is consisted of 1 vol% CO (balanced in air) and the flow rate of the reactant stream is 30 cm3 min−1, equivalent to a space velocity of 36000 cm3 h−1 gcat.−1. Reaction effluent is separated by a carbon molecular sieve (TDX-01) column and analyzed online by a gas chromatography equipped with a methanation converter and a flame ion detector. The conversion of CO can be calculated as follows:“[CO]in” and “[CO]out” mean inlet and outlet gaseous stream concentration, respectively.
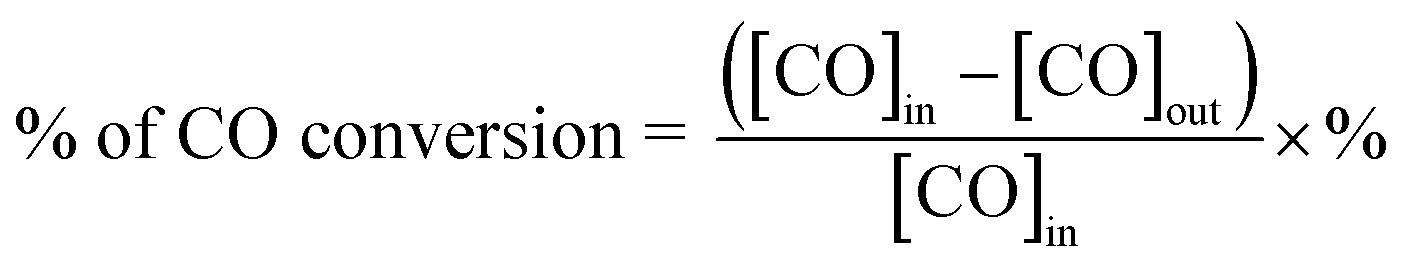
2.3 Characterization techniques
The specific areas of samples are obtained at −196 °C using a Coulter Omnisorp 100 CX. Prior to the measurement, samples are pretreated at 250 °C for 2 h in N2. X-ray powder diffraction (XRD) patterns are recorded on a Rigaku D/Max 2550PC powder diffractometer using nickel-filtered Cu Kα radiation. HRTEM images are obtained using a JEOL JEM-2110 system operating at 200 kV. H2 temperature programmed reduction (TPR) is carried out using a conventional reactor equipped with TCD. 50 mg catalyst is loaded with a flow rate of 30 ml min−1 5% H2/95% Ar. The heating rate is 10 °C min−1 from 30 °C to 800 °C. X-ray photoelectron spectra (XPS) are recorded with a Perkin-Elmer PHI-5000C ESCA spectrometer using Al Kα radiation (1486.6 eV). The base pressure is 1 × 10−8 Torr. The binding energies are calibrated using C 1s peak of contaminant carbon (BE = 284.6 eV) as standard, and quoted with a precision of ±0.2 eV. The surface composition of the samples in terms of atomic ratios is calculated, and Gaussian–Lorentzian and Shirley background were applied for peak analysis.3. Results
3.1. Catalytic CO oxidation
As shown in Fig. 1(A), the catalytic performance of CuO–CoOx–CeO2 for CO oxidation reaction is investigated. Typically, CO conversion over these ternary catalysts increases with the elevation of reaction temperature.25 Except CCC(1.2), the catalysts have a very sharp change of CO conversion. Namely, with respect to these catalysts, their temperature windows of CO conversion from 20% to 80% are only about 20 °C. That means the catalytic performance of the above catalysts is very sensitive to the reaction temperature, which has been attributed to the ability in breaking metal–oxygen bonds and the properties of the ternary catalysts.20,25 Combined with the above fact, it is reasonable to deduce that the pH value during the preparation process of the catalysts may insert an important role in the catalytic performance of the catalysts. | ||
| Fig. 1 Catalytic performance of the catalysts for CO oxidation (A) and the recycle times of CCC(1.8) catalyst (B). Reaction condition: catalyst 50 mg, flow rate 30 ml min−1, 1 vol% CO balance in air. | ||
In addition, when the relationship between the pH values and the activity of the catalysts is studied, it can be found that when the pH values increase with the addition of NaOH during the precipitation process, the activity of the corresponding catalysts for CO conversion presents a volcano curve. That is, CCC(1.2) has the lowest activity and CCC(1.8) possesses the highest CO conversion. If the catalyst activity is evaluated by the reaction temperature (T50) at which CO conversion reaches 50%, T50 over CCC(1.2) is 122 °C, but T50 over CCC(1.8) is 94 °C. It is worth to note that as for CCC(1.9), T50 sharply decreases to 106 °C in comparison with CCC(1.8) when NaOH mass increases from 1.8 g to 1.9 g but with almost identical pH value of 12.9. Obviously, additional NaOH may have an influence on the physicochemical properties of the catalyst but not the pH value.
As report that metal oxides with high thermal stability are well suited for use as catalysts and catalyst supports.30 Herein, the stability tests are carried out on CCC(1.8) catalyst, which shows the best catalytic performance. From Fig. 1(B), it is found that the conversion of CO of CCC(1.8) sample can remain the similar result when it is reused for four times, indicating the rather good stability of the ternary catalysts.
3.2. BET surface areas
N2 adsorption/desorption isotherms of the catalysts are displayed in Fig. 2. From the isotherms of the samples, the onset of the relative pressure in the hysteresis loop increase in the turn of CCC(1.2), CCC(1.4), CCC(1.6) and CCC(1.8), except that the corresponding relative pressure of CCC(1.9) is lowered once again.29 According to the BET method, the physical properties of the catalysts are listed in Table 1. Obviously, CCC(1.6) has the largest surface area (131.8 m2 g−1) among all the catalysts and CCC(1.8) comes the second (127.6 m2 g−1). But the average pore size of CCC(1.8) is 18.8 nm, much larger then those of the others, as well as the total pore volume of CCC(1.8) (0.60 cm3 g−1). Combined with the activity results in part 3.1, it comes to the conclusion that larger pore sizes may benefit to improve the diffusion of reactants to their reactive centers and enhance the activity of the catalyst.30–32 It is evident that pH have a crucial influence on the structural and surface properties of the catalysts, which may determine the catalytic performance of the catalysts in the end. | ||
| Fig. 2 N2 adsorption/desorption isotherms. | ||
3.3. XRD characterization
The XRD patterns of the catalysts are presented in Fig. 3. The characteristic peaks of CeO2 with fluorite structure are observed at 29.0°, 33.3°, 48.0°, 56.7°, 59.8°, 69.7° and 77.0°, corresponding to the standard spectrum diagram of cerianite (space groups: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225), JCPDS no. 34-0394).25,26 And the characteristic peak of Co3O4 can also be found at 31.4°, 37.0°, 38.7°, 45.0° and 65.6°, which is matched well with the standard spectrum diagram of cobalt oxide (space groups: Fdm (227), JCPDS no. 65-3103).15,30 Moreover, there is no new peak to appear or any peak to shift. Apparently, as for these catalysts, crystalline CeO2 and Co3O4 coexist to work as the support. Generally, Co atom at the interface between CeO2 and Co3O4 may diffuse into the crystalline of CeO2, which cause to the high activity of the oxygen located in Co–O–Ce bonds.25 However, in these catalysts, most are the bulk CeO2 and Co3O4. In addition, the peak of crystalline CuOx cannot be observed from the XRD pattern of samples. This indicates that CuOx are likely present in an amorphous state and/or a relatively high dispersion. However, for CCC(1.9), a peak of CuO is appeared at 35.7° (copper oxide, space groups: C2/c (15), JCPDS no. 65-2309).26 This indicates the growth of CuO particles. As the literature report,33 well-dispersed CuOx is beneficial to enhance the catalytic performance of the catalysts and the large CuO may not benefit to improve the activity of CCC(1.9).
m (225), JCPDS no. 34-0394).25,26 And the characteristic peak of Co3O4 can also be found at 31.4°, 37.0°, 38.7°, 45.0° and 65.6°, which is matched well with the standard spectrum diagram of cobalt oxide (space groups: Fdm (227), JCPDS no. 65-3103).15,30 Moreover, there is no new peak to appear or any peak to shift. Apparently, as for these catalysts, crystalline CeO2 and Co3O4 coexist to work as the support. Generally, Co atom at the interface between CeO2 and Co3O4 may diffuse into the crystalline of CeO2, which cause to the high activity of the oxygen located in Co–O–Ce bonds.25 However, in these catalysts, most are the bulk CeO2 and Co3O4. In addition, the peak of crystalline CuOx cannot be observed from the XRD pattern of samples. This indicates that CuOx are likely present in an amorphous state and/or a relatively high dispersion. However, for CCC(1.9), a peak of CuO is appeared at 35.7° (copper oxide, space groups: C2/c (15), JCPDS no. 65-2309).26 This indicates the growth of CuO particles. As the literature report,33 well-dispersed CuOx is beneficial to enhance the catalytic performance of the catalysts and the large CuO may not benefit to improve the activity of CCC(1.9).
 | ||
| Fig. 3 XRD patterns of the catalysts. | ||
The particle sizes of the metal oxides are obtained via the following formula:
| D(h k l) = Kλ/βcosθ |
3.4. HRTEM characterization
The HRTEM images of catalysts are illustrated in Fig. 4, which show the existence of CeO2 and Co3O4. For ceria particle, the reflections with d spacing values of 0.31 nm, 0.27 nm and 0.16 nm are attributed to the CeO2 (111) plane, CeO2 (200) plane and CeO2 (311) plane, respectively.36,37 And the reflections with d spacing values of 0.28 nm, 0.24 nm and 0.16 nm are belong to the Co3O4 (220) plane, Co3O4 (311) plane and Co3O4 (511) plane, respectively. However, it is interesting to find that the particle sizes of the catalysts decrease following the turn of CCC(1.2) (49.3 nm), CCC(1.9) (8.2 nm), CCC(1.4) (8.1 nm), CCC(1.6) (7.1 nm) and CCC(1.8) (6.8 nm) from the normal quantile plot as shown in Fig. 4. Hereafter, pH values during the preparation process have an essential effect on the morphology, the particle sizes and the activity of the catalysts for CO oxidation. | ||
| Fig. 4 HRTEM images and the particle size distribution of the CCC(1.2) (a and f), CCC(1.4) (b and g), CCC(1.6) (c and h), CCC(1.8) (d and i) and CCC(1.9) (e and j) catalysts. | ||
3.5. TPR characterization
As for CO oxidation, the breaking of metal–oxygen bond is one of the main factors that determine the activity of metal oxides. Fig. 5 shows the H2-TPR profiles of the samples. Compared the reduction peaks of the CoOx–CeO2 and CeO2 supports in Fig. 5(B) with that of the CuO–CoOx–CeO2 catalysts (see Fig. 5(A)), there exist reduction peaks including the reduction of CuO to Cu, CoOx to Co and CeO2 to CeO2−x for the ternary samples. Normally, the reduction of CeO2 to CeO2−x takes place at temperatures higher than 500 °C, which have little direct influence on the low-temperature reaction (e.g. reaction temperature lower than 200 °C).26 However, when transition metal ions such as Cu and Co are doped in CeO2 at the interface, these metastable crystalline structures will be easier to give active oxygen than their counterpart bulk one, namely CuO, CoOx and CeO2. | ||
| Fig. 5 H2-TPR patterns of the samples. | ||
As for CoOx–CeO2 support, the doping of Co into CeO2 at the interface possess high activity for CO oxidation, which is ascribed to the easy-breaking of Co–O–Ce bond and the superior oxygen storage release capacity.25,40,41 For example, Meng40 found that the CO oxidation should take place preferentially at the interface of Co3O4–CeO2 instead of the surface of Co3O4 in the Co3O4–CeO2 catalyst with molar ratio of 1:1. And Wang's group has investigated the oxygen storage-release capacity of Co3O4–CeO2 binary catalyst, and they found that the cobalt oxides with multiple valences can promote oxygen storage release capacity.41 As a result, the reduction peak of CoOx in CoOx–CeO2 will shift to low temperature and is assigned as γ peak here. Correspondingly, the reduction peak of CuO interacted with CeO2 shifts to low temperature and is assigned as α peak. And the reduction peak of CuO unassociated with CeO2 is assigned as β peak.
As for CCC(1.2) and CCC(1.4), the peaks at 262.2 °C and 296.5 °C are both ascribed to γ peak. And the peaks at 192.0 °C and 175.0 °C are ascribed to β peak. However, with respect to CCC(1.6) and CCC(1.8), there exist three types of reduction peaks. The peaks at 252.4 °C and 254.0 °C are ascribed to γ peak. The peaks at 198.3 °C and 178.2 °C are ascribed to β peak. And the peaks at 151.5 °C and 154.8 °C are ascribed to α peak. Evidently, the appearance of α peak leads to the shift to low reduction temperature of all peaks, which also becomes the key to improve the catalytic performance of the catalysts.30 It is worth to mention that CCC(1.8) has much better catalytic performance than CCC(1.6) though α peak of CCC(1.6) is lower than that of CCC(1.8), which is generally thought to have high activity. In our work, the energy matching has been found to play an important role in determining the catalytic performance of the catalysts.33,34 Namely, as for similar catalysts, if the neighboring reduction peaks have a narrow temperature window, this type of catalyst will possess comparable higher activity than their similar one. Here, CCC(1.8) has a narrower temperature window (23.4 °C vs. 46.8 °C) between α peak and β peak than CCC(1.6). Therefore, according to this viewpoint (i.e. energy matching), it is reasonable to deduce that CCC(1.8) has a higher activity than CCC(1.6). Nevertheless, as for CCC(1.9), α peak has almost disappeared though β peak and γ peak can be found at 182.3 °C and 267.2 °C, respectively. This cause the activity of CCC(1.9) for CO oxidation to lower down, for α peak has been thought to be the key active centers.
3.6. XPS characterization
XPS spectra of Cu 2p, Co 2p and Ce 3d for CCC(1.4), CCC(1.8) and CCC(1.9) are displayed in Fig. 6, respectively. And their corresponding composition of Cu, Co and Ce species derived from XPS is listed in Table 2. According to the data in Table 2, it is interesting to discover that on the surface of the catalysts, Co contents increase and Ce contents decrease with the elevation of pH value in the preparation step of the catalysts. The surface Cu contents of the catalysts decrease with the increase of the pH value. However, CCC(1.8) has the lowest surface Cu content. Obviously, pH value during the preparation process exerts a crucial role in the properties of the catalysts. | ||
| Fig. 6 XPS spectra of CCC(1.2), CCC(1.4) and CCC(1.8). | ||
| Sample | Binding energy (eV) | Cu/(Cu + Co + Ce)a (at%) | Co/(Cu + Co + Ce) (at%) | Ce/(Cu + Co + Ce) (at%) | ||||
|---|---|---|---|---|---|---|---|---|
| Co 2p3/2 | Co 2p1/2 | ΔE | Cu | Cu2+/Cu | Cu+/Cu | |||
| a Calculation from the deconvolution of peaks for the kinetic energy spectra of the Auger L3VV electron of samples. | ||||||||
| CCC(1.4) | 779.1 | 794.1 | 15.0 | 22.5 | 73.1 | 26.9 | 14.5 | 63.0 |
| CCC(1.8) | 779.4 | 795.4 | 16.0 | 18.5 | 29.8 | 70.2 | 29.5 | 52.0 |
| CCC(1.9) | 778.5 | 793.6 | 15.1 | 19.2 | 100 | 0 | 33.5 | 47.3 |
For Cu 2p3/2, the peak at 931.3 eV, as shown in Fig. 6(a), is attributed to Cu+/Cu2O while the peak greater than 934.4 eV is one XPS characteristic peak of Cu2+/CuO.22 Compared with CCC(1.4) and CCC(1.8), XPS peaks of CCC(1.9) shift toward higher binding energy. It is apparent that CuOx species presented in the as-prepared catalysts are different, which may be attributed to the influence of the solution pH values during the preparation process. Moreover, it can also be found that CCC(1.8) has comparable lower Cu content and higher Cu+ content on the surface of the catalysts than the other two. Combined with the data in Table 2 and Fig. 6, it is reasonable to deduce that the increase of pH value during the preparation of the catalysts may benefit the formation of Cu+ at the beginning. However, the further increase of pH value results in the oxidation of Cu+ to Cu2+, which may hinder the enhancement of the catalytic performance of the catalyst.
In Fig. 6(b), the XPS curves of Co 2p show two major peaks at ca. 779.4 and 795.4 ev, respectively.21 Generally, spin orbit split energy of Co 2p (ΔE) is associated with the oxidation state of Co. When ΔE is 16.0 eV, it is CoO species.28–35 As for Co2O3 and Co3O4, ΔE is 15.0 eV and 15.2 eV, respectively.21,28 According to the Table 2, the ΔE of the Co 2p spectra of CCC(1.8) is 16.0 eV, which attributed to CoO phase.28 At the same time, with respect to CCC(1.6) and CCC(1.9), their ΔE of the Co 2p spectra are 15.0 and 15.1 eV, respectively. This means CCC(1.6) and CCC(1.9) contain more Co2O3, which may explain the conversion of Cu+ to Cu2+ in these catalysts due to their identical surrounding.
Fig. 6(c) illustrates the oxidation states of Ce obtained from XPS measurements and analyzed by fitting the curves of Ce 3d spectra. According to the literature,18 the curves of Ce 3d spectra are composed of eight peaks corresponding to four pairs of spin–orbit doublets. Letters v and u refer to the 3d5/2 and 3d3/2 spin–orbit components, respectively. The peaks marked as v (ca. 882.4 eV), v′′ (ca. 888.2 eV) and v′′′ (ca. 898.0 eV) result from Ce4+ 3d5/2 while the peaks marked as u (ca. 900.1 eV), u′′ (ca. 907.5 eV) and u′′′ (ca. 916.3 eV) result from Ce4+ 3d3/2, and the peaks signed as v′ (ca. 886.0 eV) and u′ (ca. 904.0 eV) result from Ce3+.30 Ce3+ is created by interaction between cerium dioxide and its surrounding low valence Co and Cu atoms. The interaction between cerium oxide and cobalt oxide could lead to the inside of the ceria lattice oxygen transfer to cobalt oxide lattice and keeping the cobalt ion state at a high price, which may result in the higher catalytic performance of the ternary catalysts.30
4. Conclusions
In this study, ternary catalysts have been synthesized and the influence of precipitant pH value on the properties of the catalysts is investigated with the techniques such as BET, XRD, TPR and XPS. The results show that CCC(1.8) exhibits the best catalytic activity for CO oxidation reaction. T50 of CCC(1.8) is only 94 °C, much lower than the other catalysts. Furthermore, it is found that pH may influence not only the physical properties such as surface area and particle sizes of the catalysts but also the chemical properties such as the valence state of the elements on the surface of the catalysts. There exist more Cu+ in CCC(1.8) due to the synergistic effect between CuOx and the support such as CoOy and CeO2. This may facilitate the redox recycle of Cu2+ to Cu+ and the improvement of the catalytic performance.37–39Acknowledgements
The authors gratefully acknowledge the financial support from National Natural Science Foundation of China (nos. 21103045, 1210040, 1103312) and the Fundamental Research Funds for the Central Universities.Notes and references
- C. Ratnasamy and J. P. Wagner, Catal. Rev.: Sci. Eng., 2009, 51, 325 CAS.
- S. Carrettin, P. Concepción, A. Corma, J. M. L. Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538 CrossRef CAS PubMed.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. M. Reddy, RSC Adv., 2014, 4, 11322 RSC.
- Z. Liu, Z. Wu, X. Peng, A. Binder, S. Chai and S. Da, J. Phys. Chem. C, 2014, 118, 27870 CAS.
- S. Zhao, Y. Chen and Z. Liu, RSC Adv., 2015, 5, 33299 RSC.
- X. Guo, D. Shen, Y. Li, M. Tian, Q. Liu, C. Guo and Z. Liu, J. Mol. Catal. A: Chem., 2011, 351, 174 CrossRef CAS PubMed.
- S. A. C. Carabineiro, N. Bogdanchikova, P. B. Tavares and J. L. Figueiredo, RSC Adv., 2012, 2, 2957 RSC.
- E. Y. Ko, E. D. Park, K. W. Seo, H. C. Lee, D. Lee and S. Kim, Catal. Today, 2006, 116, 377 CrossRef CAS PubMed.
- Z. Liu, R. Zhou and X. Zheng, J. Mol. Catal. A: Chem., 2006, 255, 103 CrossRef CAS PubMed.
- E. Simsek, S. Oezkara, A. E. Aksoylu and Z. I. Onsan, Appl. Catal., A, 2007, 316, 169 CrossRef CAS PubMed.
- C. Pedrero, T. Waku and E. Iglesia, J. Catal., 2005, 233, 242 CrossRef CAS PubMed.
- L. F. Liotta, M. Ousmane, G. D. Carlo, G. Pantaleo, G. Deganello, G. Marci, L. Retailleau and A. Giroir-Fendler, Appl. Catal., A, 2008, 347, 81 CrossRef CAS PubMed.
- H. Wang, H. Zhu, Z. Qin, F. Liang, G. Wang and J. Wang, J. Catal., 2009, 264, 154 CrossRef CAS PubMed.
- Y. Teng, H. Sakurai and A. Ueda, Int. J. Hydrogen Energy, 1999, 24, 355 CrossRef CAS.
- L. F. Liotta, G. D. Carlo, G. Pantaleo and G. Deganello, Appl. Catal., B, 2007, 70, 314 CrossRef CAS PubMed.
- D. Cameron, R. Holliday and D. Thompson, J. Power Sources, 2003, 118, 298 CrossRef CAS.
- J. Papavasiliou, G. Avgouropoulos and T. Ioannides, Appl. Catal., B, 2006, 66, 168 CrossRef CAS PubMed.
- A. Martínez-Arias, A. Hungría, G. Munuera and D. Gamarra, Appl. Catal., B, 2006, 65, 207 CrossRef PubMed.
- E. Moretti, M. Lenarda, L. Storaro, A. Talon, R. Frattini, S. Polizzi, E. Rodríguez-Castellón and A. Jiménez-López, Appl. Catal., B, 2007, 72, 149 CrossRef CAS PubMed.
- Y. Chen, B. Liaw, W. Chang and C. Huang, Int. J. Hydrogen Energy, 2007, 32, 4550 CrossRef CAS PubMed.
- G. Zhou, H. Lan, R. Song, H. Xie and Q. Du, RSC Adv., 2014, 4, 50840 RSC.
- M. P. Woods, P. Gawade, B. Tan and U. S. Ozkan, Appl. Catal., B, 2010, 97, 18173 CrossRef PubMed.
- P. Gawade, B. Bayram, A. M. C. Alexander and U. S. Ozkan, Appl. Catal., B, 2012, 128, 21 CrossRef CAS PubMed.
- A. Arango-Díaz, E. Moretti, A. Talon, L. Storaro, M. Lenara, P. Núňez and E. Rodríguez-Castellón, Appl. Catal., A, 2014, 477, 54 CrossRef PubMed.
- Y. Chen, D. Liu, L. Yang, M. Meng, J. Zhang, L. Zheng, S. Chu and T. Hu, Chem. Eng. J., 2013, 234, 88 CrossRef CAS PubMed.
- Z. Liu, R. Zhou and X. Zheng, J. Mol. Catal. A: Chem., 2007, 267, 137 CrossRef CAS PubMed.
- Z. Liu, R. Zhou and X. Zheng, J. Nat. Gas Chem., 2010, 19, 313 CrossRef CAS.
- Z. Liu, R. Zhou and X. Zheng, Int. J. Hydrogen Energy, 2008, 33, 791 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- Z. Liu, S. Chai, A. Binder, Y. Li, L. Ji and S. Dai, Appl. Catal., A, 2013, 451, 282 CrossRef CAS PubMed.
- L. F. Liotta, G. D. Carlo, G. Pantaleo and G. Deganello, Appl. Catal., B, 2007, 70, 314 CrossRef CAS PubMed.
- L. F. Liotta, G. D. Carlo, G. Pantaleo, A. M. Venezia and G. Deganello, Appl. Catal., B, 2006, 60, 217 CrossRef PubMed.
- X. Jiang, G. Lu, R. Zhou, J. Mao, Y. Chen and X. Zheng, Appl. Surf. Sci., 2001, 173, 208 CrossRef.
- Z. Liu, Y. Xie, W. Li, R. Zhou and X. Zheng, J. Nat. Gas Chem., 2011, 20, 111 CrossRef CAS.
- H. Wang, H. Q. Zhu, Z. F. Qin, F. X. Liang, G. F. Wang and J. Q. Wang, J. Catal., 2009, 264, 154 CrossRef CAS PubMed.
- H. Wang, H. Zhu, Z. Qin, G. Wang, F. Liang and J. Wang, Catal. Commun., 2008, 9, 1487 CrossRef CAS PubMed.
- Z. Liu, S. Chai, A. Binder, Y. Li, L. Ji and S. Dai, Appl. Catal., A, 2013, 451, 282 CrossRef CAS PubMed.
- Y. Chen, D. Liu, L. Yang, M. Meng, J. Zhang, L. Zheng, S. Chu and T. Hu, Chem. Eng. J., 2013, 234, 88 CrossRef CAS PubMed.
- S. Yao, K. Mudiyanselage, W. Xu, A. C. Johnston-Peck and J. C. Hanson, ACS Catal., 2014, 4, 1650 CrossRef CAS.
- J. Luo, M. Meng, X. Li, X. Li, Y. Zha, T. Hu, Y. Xie and J. Zhang, J. Catal., 2008, 254, 310 CrossRef CAS PubMed.
- J. Wang, M. Shen, J. Wang, J. Gao, J. Ma and S. Liu, Catal. Today, 2011, 175, 65 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2015 |