
A rheological study on non-rubber component networks in natural rubber
Lili
Xu
,
Cheng
Huang
,
Mingchao
Luo
,
Wei
Qu
,
Han
Liu
,
Zhewei
Gu
,
Liumei
Jing
,
Guangsu
Huang
* and
Jing
Zheng
*
College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: GuangsuHuang@scu.edu.cn; zhengjing@scu.edu.cn
First published on 24th September 2015
Abstract
Phospholipids and proteins were separately removed as the main non-rubber components to individually study their effect on the structure and properties of the rubber. Fourier Transform Infrared Spectroscopy (FTIR) and 1H nuclear magnetic resonance spectroscopy (1H-NMR) were used to characterize the chemical structure and the residual non-rubber component. A rheology study and stress relaxation measurements were used to study the role that non-rubber components play in natural networks. A rheological study showed that natural rubber (NR) and deproteinized natural rubber (DPNR) exhibited similar dynamic modulus at 170 °C. The lack of superposition in van Gurp–Palmen (vGP) curves at different temperatures for NR and DPNR, together with the shape of vGP curves, proved that long chain branching was mainly constructed by phospholipids. Stress relaxation measurements at room temperature were fitted with the Maxwell model and showed that the NR relaxation curve underwent a quick decrease and then came to an equilibrium stress retention of 58%, which is about 3 times higher than that of DPNR, indicating that proteins in NR contributed to the network structure at room temperature. Combining molecular dynamic studies, the interaction of proteins and phospholipids in non-rubber networks was proposed.
Introduction
Natural rubber (NR) has been a particularly interesting and attractive material since it was first vulcanized by C. Goodyear in 1839. Natural rubber (NR) latex, collected from Hevea trees as a colloidal suspension,1 is the main commercial feedstock for the rubber and latex dipping industries. The tire industry is one for which the applications of natural rubber are highly attractive, and obvious superiorities in mechanical and universal properties in a wide temperature range could allow such materials to be employed as innovative solutions to satisfy the demanding requirements that a modern-day tire must fulfill, such as wet grip, mechanical and thermal resistance, low cost, recyclability, etc.2,3 Nowadays, natural rubber has become increasingly important for applications spanning many fields of chemistry, the hygiene and medical sectors, and it is further involved in a broad spectrum of consumer products in our daily life.4–7As a natural material, the yield of natural rubber which is seriously restricted by land resources and natural conditions, has become the fatal bottleneck for its extensive application. Thus, the synthetic analogue polyisoprene as the best alternative, is modifed mainly by various molecular design methods,8–13 however the properties were disappointing.
It has been reported that non-rubbers,14 especially proteins and lipids, give natural rubber latex excellent properties which are unsurpassed by any synthetic latex.15 As a consequence, great industrial and scientific attention has been paid to the structures of non-rubbers and their importance on the structure of natural rubber to create the magical mechanical performance,16 such as the high strength of raw rubber (green strength), shape retention, high tensile strength, high crack growth resistance and minimal heat build up in the vulcanized state. Several attempts have also been made to study non-rubber components and investigate their corresponding structures in natural rubber. Y. Tanaka17,18 first proposed that cis-polyisoprenes obtained from H. brasiliensis and Parthenium argentatum consist of more than 5000 isoprene units and that there are trans-isoprene units per rubber chain. The mainstream view now is that NR molecules comprise 2 trans-isoprene units connected to a long chain of cis-isoprene units. Two terminal groups, referred to as α and ω, have been postulated to link with mono- and di-phosphate groups associated with the phospholipids by H-bonding at the α-terminal, whereas the ω-terminal is postulated to be a modified dimethylallyl group linked to the proteins by H-bonding.19 Then the non-rubbers can connect the linear polyisoprene chains in NR through functional terminals and generate a branching or network topology, which is designated a “naturally occurring network”.20 But since the Goodyear medal19 in 2001, no clear evidence of the structure of an end-linking network has been reported.
Although the real cross-linking structure and role of non-rubbers are not fully understood, it has been suggested that the cross-linking of NR formed by non-rubbers can be eliminated by deproteinization and transesterification. After the removal of proteins and phospholipids, linear rubber chains can be obtained.21
The effect of proteins and phospholipids on strain induced crystallization and the correlation with the tensile strength of NR have been extensively studied using X-ray diffraction and microscopic methods.22,23 However, spectroscopy methods are static and mostly accompanied by solvent, and X-ray characterization is indirect in “naturally occurring” network characterization. Rheological properties are highly sensitive to long chain branching because even a low degree of long chain branching has a significant effect on the viscoelasticity in polyethylenes23–28 or polypropylene.29 A van Gurp–Palmen curve, which was first proposed to judge the feasibility of Time–Temperature Superposition (TTS) principles and then evolved to branched structure studies independent of the chemical components of the macromolecule chain, is also very sensitive to the long chain branching of the polyisoprene melt in unvulcanized NR.30 Therefore, the rheological behavior of unvulcanized NR can be characterized using a dynamic spectrum, related to the molecular structure in the melt, which is strongly impacted on by branching or networking. However, to our knowledge, there is a lack of a systematic investigation on how non-rubber components affect the rheological behavior of rubber chains in melts and inversely testify the role of non-rubber components from rheology, especially the effect of long chain branching on rheological properties. While few rheological studies were conducted on NR, among which most focused on blended NR with plastic,31 silica,32,33 carbon black34 or another natural ingredient35 in a vulcanized regime, hardly any were on unvulcanized rubber. While for unvulcanized NR, the studies mainly focused on processability36 or nonlinear viscoelastic behavior.37 In this study, we conducted rheological studies on an unvulcanized NR melt, and introduced the widely used vGP curve to study the effect of non-rubber components on unvulcanized NR.
In addition, the stress relaxation behavior also provides important guidance in probing the network, and additionally the equilibrium relaxation modulus G∞ is proportional to the crosslinking density.28 The stress relaxation behavior of natural rubber is often investigated for vulcanized rubber,29–31 while scarcely for the unvulcanized if any. In this study, to investigate the network structure constructed by non-rubbers in unvulcanized NR, we introduced a stress relaxation test and analyzed the equilibrium relaxation modulus to evaluate the crosslinking density and the constraints on relaxation units.
In this work, an attempt was made to clarify the “naturally occurring network” through a molecular dynamics study, combining the chemical structure analysis. Fourier Transform Infrared Spectroscopy (FTIR), rheology and stress relaxation were used to analyze the composition and structure of the natural network. To individually study the effect of the non-rubber components, three models were prepared to individually study the effect of non-rubber components which are listed as follows, NR containing all of the non-rubber components, deproteined natural rubber (DPNR) containing phospholipids and fatty acids, transesterified deproteinized natural rubber (TEDPNR) containing only rubber chains.
Experimental
Materials
Natural rubber latex used in the present work was commercial high ammonia natural rubber (HANR) latex purchased from Dongfeng company (China). Total solid content and dry rubber content of the HANR latex, determined according to ASTM D 1076, were 61.3% and 60.8 w/w%, respectively. Triton X-100 and protease (P-5380) was provided by Sigma-Aldrich. Other reagents used were analytical grade.Preparation of specimens
Three models were prepared to individually study the effect of the non-rubber components which are listed as follows, natural rubber containing all of the non-rubber components, deproteined natural rubber (DPNR) containing phospholipids and fatty acids, transesterified deproteinized natural rubber (TEDPNR) containing only rubber chains.Natural rubber
HANR latex was centrifuged once for the removal of debris, and then spread into a thin layer to dry under reduced pressure at ambient temperature until it remained a constant weight in a vacuum oven.Deproteinization of natural rubber
Deproteinization of the HANR latex was carried out through incubation with 0.08 wt% protease in the presence of 0.15% v/v Triton X-100 and 1 wt% aqua ammonia at 37 °C for 12 h. The dry rubber content was 30%. The cream fraction was redispersed in the surfactant, in which the dry rubber content was adjusted to 30 wt% with deionized water. It was washed two or three times by centrifugation. The rubber was recovered by centrifugation followed by coagulation with methanol and dried under reduced pressure at ambient temperature until it remained a constant weight.Measurement of the nitrogen content of the deproteined natural rubber (DPNR) was made using a nitrogen elemental analyzer (CARLO ERBA 1106, Italian), and the nitrogen content was reduced from 0.5% to 0.07% after deproteinization.
Transesterification of the deproteined rubber
Transesterification of the rubber from DPNR was carried out in 2% w/v toluene through reaction with freshly prepared sodium methoxide and stirring at room temperature for 8 h. The resulting transesterified deproteinized natural rubber (TEDPNR) was purified by precipitation of the rubber solution using an excess of methanol and then dried in a vacuum oven.Measurement of the phosphorus content of TEDPNR was made using a phosphorus elemental analyzer (IRIS 1000 ICP-AES, Thermo Electron Co. USA), and the phosphorus content was reduced from 224 ppm to 65 ppm after transesterification.
The molecular weight of all samples was determined using gel permeation chromatography (HLC-8320, Waters), and the number averaged molecular weight was about 150–160 kg mol−1 for NR and DPNR, 134 kg mol−1 for TEDPNR, while the MW of commercial linear isoprene rubber (IR) was 317 kg mol−1.
Characterization
Results and discussion
Chemical structure characterization of the NR samples
Infrared spectroscopy was used as a method for chemical structure analysis, and also to study intermolecular interactions caused by hydrogen bonding, because the vibrational modes of the donor and acceptor groups are sensitive to this interaction leading to a change in the vibrational characteristics.32 The FTIR spectra of NR, DPNR and TEDPNR at room temperature are plotted in Fig. 1. | ||
| Fig. 1 FTIR spectra of NR, DPNR and TEDPNR. | ||
First, a wide H-bond peak at around 3400 cm−1 was absent in TEDPNR. For NR the wide peak comprised a relatively sharp peak at about 3300 cm−1 related to the N–H symmetric stretch of the H-bond, and the wide H-bond peak is mainly ascribed to the O–H symmetric stretch in the H-bond in non-rubbers, overlapping with a small amount of environmental water absorbed by non-rubbers.
Second, to pick out the non-rubbers, the assignment of the rubber chain peaks are listed below. vmax/cm−1 = 836 (trisubstituted olefin out-of-plane ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, wag), 1129 (–CH3, rock), 1300 (–CH2–, wag), 1376 (–CH3, symmetric deformation), 1450 (–CH2– symmetric and –CH3 asymmetric deformation), 1664 (CC, stretch), 2720 (overtone of –CH2– umbrella), 2850 (–CH2– and –CH3, symmetric stretch), 2920 (–CH2–, asymmetric stretch), 2962 (–CH3, asymmetric stretch), and 3030 (olefin CH– stretch).33,34
CH, wag), 1129 (–CH3, rock), 1300 (–CH2–, wag), 1376 (–CH3, symmetric deformation), 1450 (–CH2– symmetric and –CH3 asymmetric deformation), 1664 (CC, stretch), 2720 (overtone of –CH2– umbrella), 2850 (–CH2– and –CH3, symmetric stretch), 2920 (–CH2–, asymmetric stretch), 2962 (–CH3, asymmetric stretch), and 3030 (olefin CH– stretch).33,34
The recession of the characteristic protein peaks at 1663 and 1546 cm−1 (ref. 35) is observed in DPNR compared with NR, and the CO phospholipid peaks at 1738 cm−1 (ref. 36) in TEDPNR. The FTIR results, in combination with the elemental analysis in Section 2, proved that the intended samples were successfully prepared. As for the different intensity of the CC stretch peak in the normalized infrared spectra, it can be interpreted as occurring due to various infrared activities with different non-rubber environments.
The shoulder peak at 1260 cm−1 in NR was shifted to 1248 cm−1 and became sharp after protein removal, continually shifting to about 1243 cm−1 and receding rapidly after phospholipid deprivation. It has been reported that the asymmetric O–P–O vibrational band is extremely sensitive to hydration, and the O–P–O asymmetric stretching of the hydrated phospholipid bilayer is usually observed at 1230 cm−1, whereas dried or anhydrous lipid always appears at a 30 cm−1 higher frequency.37 The shift of the O–P–O asymmetric stretching peak strongly suggests that the phospholipids in NR interacted with the protein via H-bonds, and in DPNR rubber they aggregate or link together via H-bonds.38
1H-NMR
The 1H-NMR spectra of NR, DPNR and TEDPNR are shown in Fig. 2, and a small residual chloroform peak was detected at 7.28 ppm. All of the spectra were normalized using theC–H at the backbone triplet (J = 6.5 Hz) peak at 5.15 ppm. A small multiplet signal resonating at 3.91 ppm was expected to be derived from the nonequivalent methylene protons linked to a phosphate group, –C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2OP–.39 The multiple peaks at 2.365 ppm, 2.375 ppm and 2.395 ppm were assignable to the –CH2– of long-chain fats, the multiple peak at 2.06 ppm was assignable to –C2– in the backbone, the single peak at 1.7 ppm was assignable to –C3 in the backbone, the multiple peak at 1.43 ppm was assigned to the –C2– of protein, and the multiple peak at 1.13 ppm was assigned to the –C3 of protein. From the extended spectra we can see that protein was removed after deproteinization, long-chain fat was eliminated after transesterification, while phospholipid was partly removed in TEDPNR. This can be interpreted by the suggestion that some phospholipid was connected with the rubber chain terminal.16
2OP–.39 The multiple peaks at 2.365 ppm, 2.375 ppm and 2.395 ppm were assignable to the –CH2– of long-chain fats, the multiple peak at 2.06 ppm was assignable to –C2– in the backbone, the single peak at 1.7 ppm was assignable to –C3 in the backbone, the multiple peak at 1.43 ppm was assigned to the –C2– of protein, and the multiple peak at 1.13 ppm was assigned to the –C3 of protein. From the extended spectra we can see that protein was removed after deproteinization, long-chain fat was eliminated after transesterification, while phospholipid was partly removed in TEDPNR. This can be interpreted by the suggestion that some phospholipid was connected with the rubber chain terminal.16
 | ||
| Fig. 2 (a) 1H-NMR spectra of NR, DPNR and TEDPNR in the range of 0–7.5 ppm, extended 1H-NMR spectra of NR, DPNR and TEDPNR normalized using the 5.15 ppm peak in the range of (b) 4.9–5.4 ppm, (c) 1.10–1.16 ppm, (d) 1.39–1.47 ppm, (e) 3.80–4.00 ppm, and (f) 2.34–2.42 ppm. | ||
Combining the 1H-NMR and FTIR results, we can conclude that the intended samples were successfully prepared.
Branched molecular structure in unvulcanizated natural rubber
To further study the effect of non-rubbers on the main chain structure, the rheological behavior is analyzed in this Section.All measurements were conducted at a fixed strain, ranging from 2% to 4% at 170 °C where all samples display a linear viscoelastic performance, and the storage modulus G′ as a function of frequency is shown in Fig. 3. At such a high temperature, most H-bonding or other non-bonding interactions were destroyed at this temperature.
 | ||
| Fig. 3 Dynamic spectra of NR, DPNR and TEDPNR at 170 °C. | ||
NR and DPNR show a higher modulus at low frequency than TEDPNR, indicating the existence of long-chain branching, crosslinking or phase-separation.40,41 However, the modulus of NR is a little higher than that of DPNR. DPNR displayed no apparent slope change at low frequency, while the modulus increased by as much as 2 orders of magnitude in contrast with TEDPNR.
To further investigate the modulus change, dynamic spectra at different temperatures were obtained for all of the samples, and van Gurp–Palmen plots were drawn using dynamic shear modulus values at three different temperatures (160, 170, and 190 °C), displayed in Fig. 4. Generally, very good superposition of the vGP curves was observed for classical viscoelastic polymers, while a small amount of long chains can significantly distort the vGP curve and thus cause a misalignment.42,43 The rubber models were confirmed to be thermo-rheological melts from the discrepancy of the vGP curves for NR and DPNR in Fig. 4, possibly with long-chain branching or phase-separation.
 | ||
| Fig. 4 vGP curves for (a) NR, (b) DPNR and (c) TEDPNR at various temperatures. | ||
To judge the reason for the discrepancy in the vGP curves, a temperature sweep from 100 °C to 200 °C at a frequency of 1 Hz at 1% strain was conducted. As is known, phase-separation can cause a sharp uprush in the modulus of the temperature sweep.44,45 But here, the results show no uprush in the dynamic modulus in Fig. 5, indicating that no phase separation occurred in the testing temperature region. As is shown in Section 2, all samples were unvulcanized, so we can exclude crosslinking. After excluding phase-separation, the modulus shift of NR and DPNR in Fig. 3 is attributed to long chain branching, and a small amount of protein involved in forming long-chain branches.
 | ||
| Fig. 5 Temperature sweep at a frequency of 1 Hz at 1% strain from 100 °C to 200 °C. | ||
In contrast to NR and DPNR, the overlap of the vGP curves at the various temperatures is good for TEDPNR, indicating that TEDPNR is a simple fluid. From the shape of the curve – only one minimum, spread monotonically to 90° in the phase angle – we can be conclude that TEDPNR was probably linear. Combining the good linearity of the Han map46 in Fig. 6, we can conclude that the curve for TEDPNR was linear at high temperature, in accordance with the dynamic spectrum.
 | ||
| Fig. 6 Han map of DPNR and TEDPNR at 170 °C. | ||
The above rheological results showed that long-chain branching existed only in the NR and DPNR melt, indicating that phospholipids and a small amount of protein acted as the branching point in the long-chain branching construction. Furthermore, we can conclude that phospholipids and a small amount of protein connected with the rubber terminal point by chemical bonds or very strong interactions (which survive at 170 °C), then the non-rubbers coagulate through micellar interaction,47 and possibly formed the star structure in the melt.48
Network structure in unvulcanizated natural rubber
Rheological experiments on the structure of the three models at high temperature confirms the stable topology in NR, which proves in turn that the natural network is further constructed mainly through thermally unstable H-bonding or non-bonding interactions between the non-rubber components and rubber chain ends, in accordance with the FTIR results. However, does the natural network exist in melts at room temperature? To answer this question, stress relaxation tests were conducted. As is known, stress relaxation analysis is an effective method for studying the network, and the equilibrium relaxation modulus G∞ is proportional to the crosslinking density.44In this study, the stress relaxation curves of NR, DPNR and TEDPNR at 25 °C are displayed in Fig. 7, where all of the samples were quickly subjected to a strain of 20% to avoid the influence of strain induced crystallinity (SIC),49 and then the strain was maintained for 1200 s. For the sake of contrast, a relaxation curve of fairly linear IR with a 317 kg mol−1 average molecule weight (MW) was added. Generally, the stress of linear viscoelastic polymers quickly relaxed to a very small value, and even to zero.
 | ||
| Fig. 7 Stress relaxation curves of NR, DPNR, TEDPNR and IR at 25 °C. | ||
To illustrate the effect of non-rubbers on the stress relaxation tendency, all of the instant stress values were normalized using the corresponding initial stress, as is displayed in Fig. 7. The relaxation curves of NR tended to form a plateau after about 300 s and the retention is more than 50%, the largest stress retention, indicating that protein mainly contributed to the natural network at room temperature. Unexpectedly, DPNR produced nearly the same behavior as IR, but relaxed slightly slower than linear IR. TEDPNR relaxed to zero during the testing period. It can be confirmed that the relaxation units in NR were obviously confined by a chemical or strong physical network. While in DPNR, in contrast to NR the network constraint was scarcely observed. However, in comparison with linear IR, we can conclude that the restriction derived from long chain branching. TEDPNR displayed typical relaxation behavior of linear molecular chains with fully relaxed stress.
To further confirm the above assumption, we adopted the Maxwell model41,50,51 to describe the stress relaxation of rubber, as shown in eqn (1):
 | (1) |
In eqn (1), σ(t) represents the stress in the relaxation, σe is the equilibrium stress, σi is the coefficient of the ith Maxwell model and τi is the relaxation time of the ith Maxwell model.
Elastomer relaxation followed a 7-element Maxwell model,52 as shown in eqn (2):
 | (2) |
Divided by the initial stress σ0 on both sides, eqn (2) may then be inverted to read:
 | (3) |
Let
 | (4) |
Then we fitted the experimental data from eqn (3) and the results are listed in Table 1.
| Sample | A 0 | A 1 | T 1 | A 2 | t 2 | A 3 | t 3 | R 2 |
|---|---|---|---|---|---|---|---|---|
| NR | 0.58507 | 0.06667 | 9.87772 | 0.13743 | 57.85191 | 0.18798 | 364.7487 | 0.9876 |
| DPNR | 0.15905 | 0.09697 | 12.62921 | 0.20228 | 82.65005 | 0.51581 | 544.9699 | 0.99534 |
| IR | 0.06292 | 0.13534 | 17.76458 | 0.27497 | 96.31696 | 0.50153 | 561.9759 | 0.99674 |
| TEDPNR | −0.09715 | 0.21816 | 16.88401 | 0.33616 | 115.2694 | 0.48798 | 740.3617 | 0.9829 |
From eqn (4), we can see that Ai represent the contribution of ti, and ti corresponds to the different relaxation unit in the samples. The values of A1, A2 and A3 increased in the same samples with increasing the magnitude of the relaxation times t1, t2 and t3. The consistency of Ai and ti proved that the larger relaxation unit contributed to larger stress relaxation.
Second, the crosslinking density was discussed through A0. As is shown in Table 1, NR displays the largest A0, quadruple that of DPNR, while the A0 of DPNR is also higher than that in linear IR, and TEDPNR shows a meaningless negative A0.
From eqn (4) we can see that A0 represents the stress retention rate, which is proportional to the equilibrium relaxation modulus G∞ and crosslinking density, regardless of being chemical or physical. Then we can conclude that NR possessed the largest cross-linking density, and the long chain branches in DPNR constructed a small amount of a strong entanglement network. From the crosslinking density value which is 3 times higher, we can also conclude that part of the network structure in NR is connected by interaction between proteins. Combined with the FTIR study, we can conclude that the interaction between proteins and phospholipids can also contribute to the natural network, in accordance with previous studies.53,54
For the linear IR, the super-high MW contributed to a strong entanglement network, but was still weaker than in DPNR. While in TEDPNR, the linear topology and lower MW results in the absence of a strong entanglement network.
Then the relaxation time was also considered. The relaxation time of all of the samples is displayed in the histogram in Fig. 8.
 | ||
| Fig. 8 Maxwell relaxation time histogram of NR, DPNR, TEDPNR and IR. | ||
As is shown in Fig. 8, the relaxation times decrease in this order: NR < DPNR < IR < TEDPNR. In vulcanized rubber, the relaxation time decreases with increasing crosslinking density, which can be interpreted as the crosslinking point restriction.38 Similarly, in the unvulcanized rubber, the relaxation units were also constrained by the natural network, and then displayed a decrease in relaxation time. The results confirmed that the crosslinking density increased in the order: NR > DPNR > IR > TEDPNR, which agreed with the previous study of A0.
From the above results, we proposed the non-rubber in the structures of NR and DPNR shown in Scheme 1. In NR, the protein is connected in a ω-terminal and then coagulated with each other through H-bonds, part of which can also interact with phospholipids through H-bonds. Moreover, in the α-terminal, mono- or di-phosphate is bonded with the rubber chain, and then connected by a micelle cluster. While in DPNR, the phospholipids or protein fragments after the protease treatment can also interact with the ω-terminals by polar–polar interactions or H-bonds, together with the α-terminals being connected to phospholipids to form the network, whose density is much lower than that in NR.
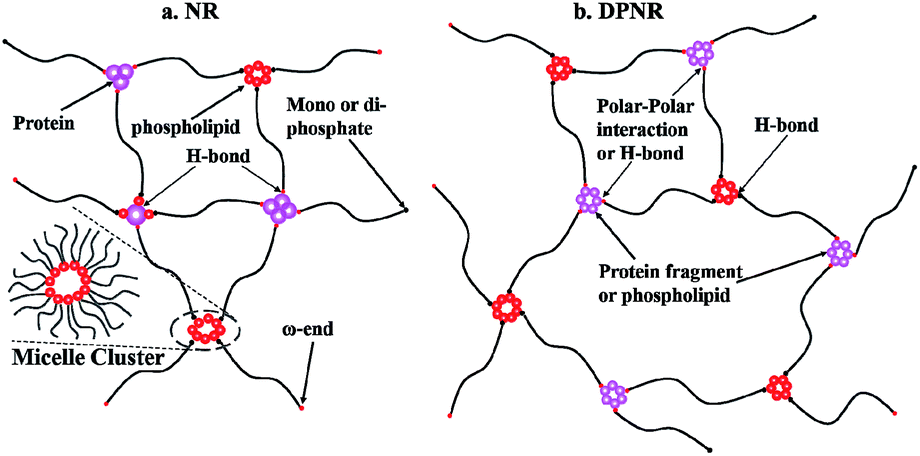 | ||
| Scheme 1 Assumed network structure in (a) NR and (b) DPNR. | ||
Conclusions
The long chain branching points formed by phosphates, and crosslinking points consisting of proteins and phospholipids are confirmed through the rheological and stress relaxation changes of the three models. The modulus decreased at low frequency, and the equilibrium stress retention decreased by 75% after deproteinization, suggesting that the network was broken through removing the protein. The van Gurp–Palmen curves showed a very good overlap and the stress relaxed to zero after transesterification, indicating the decomposition of long chain branching points by phosphatase. The FTIR study of NR and DPNR further reveals that the H-bonding between proteins and phosphatase connected with the terminal phosphatide mainly contributed to the physical crosslinking in NR. While in DPNR, some of the phospholipids and protein fragments after the protease treatment may also be connected with the ω-terminals by H-bonds to form the weak network, and H-bonds between phosphatase and α-terminal phosphatide participated together in the formation of long chain branching points.Acknowledgements
We acknowledge funding from the national natural science foundation of China to carry out this work (contract grant number 51333003). Authors must particularly acknowledge GS Huang for her forceful guidance and generous assistance.Notes and references
- G. Verhaar, Natural latex as a colloidal system, Rubber Chem. Technol., 1959, 32(5), 1627–1659 CrossRef.
- K. Mitarai, Kneading carbon black and silica to a discharging temperature of 130 degrees c. or higher; nonhardening on storage, US. Pat., 6,743,853, 2004.
- X. Li, et al., Experimental study on the temperature dependence of hyperelastic behavior of tire rubbers under moderate finite deformation, Rubber Chem. Technol., 2011, 84(2), 215–228 CrossRef CAS.
- N. Bitinis, et al., Structure and properties of polylactide/natural rubber blends, Mater. Chem. Phys., 2011, 129(3), 823–831 CrossRef CAS PubMed.
- K. Wrangsjö, et al., Primary prevention of latex allergy in healthcare—spectrum of strategies including the European glove standardization, Contact Dermatitis, 2012, 66(4), 165–171 CrossRef PubMed.
- D. Quitmann, et al., Solvent-sensitive reversible stress–response of shape memory natural rubber, ACS Appl. Mater. Interfaces, 2013, 5(9), 3504–3507 CAS.
- N. Elango and A. Faudzi, A review article: investigations on soft materials for soft robot manipulations, Int. J. Adv. Des. Manuf. Technol., 2015, 1–11 Search PubMed.
- V. Jitchum and S. Perrier, Living radical polymerization of isoprene via the RAFT process, Macromolecules, 2007, 40(5), 1408–1412 CrossRef CAS.
- E. Schoenberg, et al., Polyisoprene, Rubber Chem. Technol., 1979, 52(3), 526–604 CrossRef CAS.
- E. Schoenberg, D. Chalfant and R. Mayor, Preformed Aluminum Triisobutyl-Titanium Tetrachloride Catalysts for Isoprene Polymerization, Rubber Chem. Technol., 1964, 37(1), 103–120 CrossRef.
- S. Kaita, et al., An efficient gadolinium metallocene-based catalyst for the synthesis of isoprene rubber with perfect 1,4-cis microstructure and marked reactivity difference between lanthanide metallocenes toward dienes as probed by butadiene-isoprene copolymerization catalysis, Macromolecules, 2004, 37(16), 5860–5862 CrossRef CAS.
- Z. Yuan and M. Gauthier, Synthesis of arborescent isoprene homopolymers, Macromolecules, 2005, 38(10), 4124–4132 CrossRef CAS.
- D. Uhrig and J. W. Mays, Synthesis of combs, centipedes, and barbwires: poly(isoprene-graft-styrene) regular multigraft copolymers with trifunctional, tetrafunctional, and hexafunctional branch points, Macromolecules, 2002, 35(19), 7182–7190 CrossRef CAS.
- L. Tarachiwin, Y. Tanaka and J. Sakdapipanich, Structure and origin of long-chain branching and gel in natural rubber, Kautsch. Gummi Kunstst., 2005, 58(3), 115–122 CAS.
- H. Hasma and A. Subramaniam, Composition of lipids in latex of Hevea brasiliensis clone RRIM 501, J. Nat. Rubber Res., 1986, 1, 30–40 CAS.
- Y. Tanaka, Structural characterization of naturally occurring cis- and trans-polyisoprenes by 13C-NMR spectroscopy, J. Appl. Polym. Sci.: Appl. Polym. Symp., 1989, 44, 1–9 CAS.
- Y. Tanaka, et al., Initiation of biosynthesis in cis polyisoprenes, Phytochemistry, 1995, 39(4), 779–784 CrossRef CAS.
- L. Tarachiwin, et al., Structural characterization of α-terminal group of natural rubber. 2. Decomposition of branch-points by phospholipase and chemical treatments, Biomacromolecules, 2005, 6(4), 1858–1863 CrossRef CAS PubMed.
- Y. Tanaka, Structural characterization of natural polyisoprenes: solve the mystery of natural rubber based on structural study, Rubber Chem. Technol., 2001, 74(3), 355–375 CrossRef CAS.
- S. Toki, et al., Multi-scaled microstructures in natural rubber characterized by synchrotron X-ray scattering and optical microscopy, J. Polym. Sci., Part B: Polym. Phys., 2008, 46(22), 2456–2464 CrossRef CAS PubMed.
- J. Tangpakdee and Y. Tanaka, Characterization of sol and gel in Hevea natural rubber, Rubber Chem. Technol., 1997, 70(5), 707–713 CrossRef CAS.
- J. Carretero-Gonzalez, et al., Real-time crystallization of organoclay nanoparticle filled natural rubber under stretching, Macromolecules, 2008, 41(7), 2295–2298 CrossRef CAS.
- J. Vega, et al., Small-amplitude oscillatory shear flow measurements as a tool to detect very low amounts of long chain branching in polyethylenes, Macromolecules, 1998, 31(11), 3639–3647 CrossRef CAS.
- H. W. Shen, et al., Rheological behaviors and molecular weight distribution characteristics of bimodal high-density polyethylene, J. Appl. Polym. Sci., 2011, 121(3), 1543–1549 CrossRef CAS PubMed.
- P. M. Wood-Adams and J. M. Dealy, Using rheological data to determine the branching level in metallocene polyethylenes, Macromolecules, 2000, 33(20), 7481–7488 CrossRef CAS.
- V. C. Hugo Rolón-Garrido, M. Zatloukal and M. H. Wagner, Increase of long-chain branching by thermo-oxidative treatment of LDPE: chromatographic, spectroscopic, and rheological evidence, J. Rheol., 2013, 57(1), 105 CrossRef.
- V. H. Rolón-Garrido, J. Luo, and M. H. Wagner, Increase of Long-chain Branching by Thermo-oxidative Treatment of LDPE, in NOVEL TRENDS IN RHEOLOGY IV, AIP Publishing, 2011 Search PubMed.
- P. D. Iedema, et al., Development of MWD and branching during peroxide modification of High-Density Polyethylene by SEC-MALS and Monte Carlo simulation, Polymer, 2013, 54(16), 4093–4104 CrossRef CAS PubMed.
- F. Zulli, et al., Rheology of long-chain branched polypropylene copolymers, J. Appl. Polym. Sci., 2013, 127(2), 1423–1432 CrossRef CAS PubMed.
- S. Trinkle, P. Walter and C. Friedrich, van Gurp–Palmen plot II–classification of long chain branched polymers by their topology, Rheol. Acta, 2002, 41(1–2), 103–113 CrossRef CAS.
- C. Nakason, Y. Panklieng and A. Kaesaman, Rheological and thermal properties of thermoplastic natural rubbers based on poly (methyl methacrylate)/epoxidized-natural-rubber blends, J. Appl. Polym. Sci., 2004, 92(6), 3561–3572 CrossRef CAS PubMed.
- F. Zhang, et al., Network evolutions in both pure and silica-filled natural rubbers during cyclic shear loading, RSC Adv., 2014, 4(51), 26706 RSC.
- A. Meera, et al., Nonlinear viscoelastic behavior of silica-filled natural rubber nanocomposites, J. Phys. Chem. C, 2009, 113(42), 17997–18002 CAS.
- J. L. Leblanc, Large amplitude oscillatory shear experiments to investigate the nonlinear viscoelastic properties of highly loaded carbon black rubber compounds without curatives, J. Appl. Polym. Sci., 2008, 109(2), 1271–1293 CrossRef CAS PubMed.
- J. A. Byars and L. Jong, Flow properties of natural rubber composites filled with defatted soy flour, J. Appl. Polym. Sci., 2009, 111(4), 2049–2055 CrossRef CAS PubMed.
- C. Kim, et al., Better characterization of raw natural rubber by decreasing the rotor speed of Mooney viscometer: role of macromolecular structure, Polym. Eng. Sci., 2010, 50(2), 240–248 CAS.
- J. L. Leblanc, Nonlinear viscoelasticity of (unvulcanized) natural rubber, derived materials, and compounds through LAOS testing, Rubber Chem. Technol., 2010, 83(1), 65–96 CrossRef CAS.
- A. Batra, C. Cohen and L. Archer, Stress relaxation of end-linked polydimethylsiloxane elastomers with long pendent chains, Macromolecules, 2005, 38(16), 7174–7180 CrossRef CAS.
- L. Tarachiwin, et al., Structural characterization of alpha-terminal group of natural rubber. 1. Decomposition of branch-points by lipase and phosphatase treatments, Biomacromolecules, 2005, 6(4), 1851–1857 CrossRef CAS PubMed.
- J. Berry and W. Watson, Stress relaxation of peroxide and sulfur vulcanizates of natural rubber, J. Polym. Sci., 1955, 18(88), 201–213 CrossRef CAS PubMed.
- S. A. Baeurle, A. Hotta and A. A. Gusev, A new semi-phenomenological approach to predict the stress relaxation behavior of thermoplastic elastomers, Polymer, 2005, 46(12), 4344–4354 CrossRef CAS PubMed.
- A. V. Tobolsky, I. B. Prettyman and J. H. Dillon, Stress Relaxation of Natural and Synthetic Rubber Stocks, J. Appl. Phys., 1944, 15(4), 380 CrossRef CAS PubMed.
- A. V. Tobolsky, Stress Relaxation Studies of the Viscoelastic Properties of Polymers, J. Appl. Phys., 1956, 27(7), 673 CrossRef CAS PubMed.
- H. H. Winter, Analysis of Linear Viscoelasticity of a Crosslinking Polymer at the Gel Point, J. Rheol., 1986, 30(2), 367 CrossRef CAS.
- S. Onogi, T. Matsumoto and Y. Warashina, Rheological properties of dispersions of spherical particles in polymer solutions, Trans. Soc. Rheol., 1973, 17(1), 175–190 CrossRef CAS.
- S. G. Hatzikiriakos, Long chain branching and polydispersity effects on the rheological properties of polyethylenes, Polym. Eng. Sci., 2000, 40(11), 2279–2287 CAS.
- J. Yunyongwattanakorn, et al., Effect of gel on crystallization behavior of natural rubber after accelerated storage hardening test, J. Appl. Polym. Sci., 2007, 106(1), 455–461 CrossRef CAS PubMed.
- C. He, et al., Molecular structure of high melt strength polypropylene and its application to polymer design, Polymer, 2003, 44(23), 7181–7188 CrossRef CAS PubMed.
- S. Murakami, et al., Structural development of natural rubber during uniaxial stretching by in situ wide angle X-ray diffraction using a synchrotron radiation, Polymer, 2002, 43(7), 2117–2120 CrossRef CAS.
- B. Bernstein, E. Kearsley and L. Zapas, A study of stress relaxation with finite strain, Trans. Soc. Rheol., 1963, 7(1), 391–410 CrossRef.
- J. S. Stevenson and R. P. Kusy, Force application and decay characteristics of untreated and treated polyurethane elastomeric chains, Angle Orthod., 1994, 64(6), 455–466 CAS.
- A. Johnson and C. Quigley, A viscohyperelastic Maxwell model for rubber viscoelasticity, Rubber Chem. Technol., 1992, 65(1), 137–153 CrossRef CAS.
- K. Berthelot, S. Lecomte and Y. Estevez, et al. Rubber Elongation Factor (REF), A Major Allergen Component In Hevea brasiliensis Latex Has Amyloid Properties, PLoS One, 2012, 7(10), e48065–e48065 CAS.
- K. Berthelot, et al., Rubber particle proteins, HbREF and HbSRPP, show different interactions with model membranes, Biochim. Biophys. Acta, 2014, 1838(1 Pt B), 287–299 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |