
Regioselective synthesis of polygamma (γ) acid
Fatih Doğanab,
Kevser Temizkana and
İsmet Kaya*a
aPolymer Synthesis and Analysis Laboratory, Department of Chemistry, Çanakkale Onsekiz Mart University, Çanakkale, 17020, Turkey. E-mail: kayaismet@hotmail.com; Fax: +90 286 218 05 33; Tel: +90 286 218 00 18
bFaculty of Education, Secondary Science and Mathematics Education, Çanakkale Onsekiz Mart University, 17100, Çanakkale, Turkey
First published on 5th June 2015
Abstract
The oxidative polymerization of 6-amino-4-hydroxy-2-naphthalenesulfonic acid in an aqueous alkaline medium leads to the formation of a polygamma (γ) acid consisting of regioselective 1–4 oxazine rings. Spectral characterizations were performed by NMR, FT-IR, and UV-Vis techniques. The average molecular weight and polydispersity index of the gamma acid polymer (γ-AP) were found to be 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and 1.42, respectively. γ-AP also presented an uncommon multicolored emission behavior in dimethylsulfoxide (DMSO). When excited at 405, 480, and 532 nm, γ-AP emitted blue, green and yellow light, respectively. X-ray diffraction (XRD), differential scanning calorimetry (DSC), dynamic mechanical analyses (DMA), and transmission electron microscopy (TEM) illustrated that γ-AP is composed of semicrystalline nanofiber structures with different lengths between 10–500 nm. The kinetic parameters related to the solid state thermal decomposition of γ-AP were determined using differential isoconversional methods (Friedman and Kissenger) and integral isoconversional methods (FWO, KAS, Tang, Starink and Bosewell). By using the Tang, KAS, FWO, Starink, Bosewell, Kissenger and Friedman methods, the average values of the activation energies were calculated to be 184.4, 184.4, 185.4, 188.5, 183.6, 179.7 and 187.1 kJ mol−1, respectively. The master plot curves also suggested a diffusion-type kinetic model for the solid state thermal decomposition stage of γ-AP.
000 Da and 1.42, respectively. γ-AP also presented an uncommon multicolored emission behavior in dimethylsulfoxide (DMSO). When excited at 405, 480, and 532 nm, γ-AP emitted blue, green and yellow light, respectively. X-ray diffraction (XRD), differential scanning calorimetry (DSC), dynamic mechanical analyses (DMA), and transmission electron microscopy (TEM) illustrated that γ-AP is composed of semicrystalline nanofiber structures with different lengths between 10–500 nm. The kinetic parameters related to the solid state thermal decomposition of γ-AP were determined using differential isoconversional methods (Friedman and Kissenger) and integral isoconversional methods (FWO, KAS, Tang, Starink and Bosewell). By using the Tang, KAS, FWO, Starink, Bosewell, Kissenger and Friedman methods, the average values of the activation energies were calculated to be 184.4, 184.4, 185.4, 188.5, 183.6, 179.7 and 187.1 kJ mol−1, respectively. The master plot curves also suggested a diffusion-type kinetic model for the solid state thermal decomposition stage of γ-AP.
Introduction
Conducting polyarene systems (CPS) have received increasing scientific and technological interest in multidisciplinary fields like chemical gas sensors, light emitting diodes, conductive adhesives, electromagnetic shielding, photovoltaic and optical devices, etc.1–5 Extensive studies are currently investigating CPS due to their unique electrical, optical, optoelectrical and oxidative properties, as well as thermal and environmental stability.6–9 Among these CPS, multifunctional conjugated polymers have become the focus point for modern polymer chemistry in the synthesis of a broad variety of promising new materials because of their novel properties and potential applications. Functional groups contribute to the use of CPS as anti-microbial agents, semi-conductive, graphitic, photoresistant, antistatic and electroactive materials.10–13 Because of such characteristics, the synthesis of a novel CPS including multi active centers is of great importance for more advanced applications and is a new trend in the preparation of functional materials. However, the inadequate ability for processing because of poor solubility is the main problem for practical applications of functional CPS. The solubility is usually improved by using different types of substituted groups within CPS. To date, many synthetic strategies have been developed to prepare a new soluble functional CPS.14–16 Oxidative polymerization is an alternative process to prepare a soluble conductive polymer.17 This method (OP) has a simple, cheap and environmentally sensitive synthetic pathway, and is based on a facile procedure producing less hazardous waste. The OP method allows production of electronically and optically active, and also nanostructured, CPS. This technique, therefore, is often preferred over other polymerization techniques. However, as in other procedures, more substituted groups reduce the solubility of CPS in common solvents. For this reason, the multifunctional CPS's have rarely been reported until now. So it will be interesting to study the synthesis and properties of soluble CPS consisting of different types of functional group. This investigation reports on the preparation of soluble functional CPS by using OP without using any external templates, surfactants, stabilizers or functional dopants. In this report, the photo physical, electrochemical and thermal properties of functional CPS were also presented.Experimental section
Materials
6-Amino-4-hydroxy-2-naphthalenesulfonic acid (monomer gamma acid, γ-AM) (99%, ACS reagent), sodium hypochlorite, hydrogen peroxide (30% aqueous solution), potassium hydroxide (≥85%, ACS reagent, pellets) and hydrochloric acid (37%, AR grade) were supplied by Merck Chem. Co. (Germany). In the oxidative polymerization of γ-AM, sodium hypochlorite and hydrogen peroxide were used as oxidants. Acetonitrile, ethanol, chloroform, acetone, tetrahydrofuran, dimethylsulfoxide and other solvents were purchased from Merck Chem. Co. (Germany) and then used in the solubility experiments and for the removal of unreacted monomers. Hydrochloric acid was used as neutralization solution to precipitate the polymer. All chemical were used as received.The preparation of γ-AP
γ-AM was chemically transformed into γ-AP in the presence of various oxidants such as NaOCl, H2O2 and air by oxidative polymerization in an aqueous alkaline medium (Scheme 1).18 At the start of the reaction, the 0.001 mol γ-AM was dissolved in a 50 mL three neck flask containing aqueous KOH solution of 10%. Polymerization reactions were started by drop wise addition of NaOCl or H2O2 in a refluxing system with a thermometer and magnetic stirrer. The reactions continued up to 80 °C for 15 h. In experiments in which air oxygen was used as oxidant, the CO2 in the air was neutralized with a 20% solution of KOH before releasing the reaction medium. Reactions were stopped by the addition of an equimolar amount of HCl. The polymer particles were filtered, then washed with 50 mL of hot water to remove mineral salts and oxidants, and finally dried at 60 °C in an vacuum oven.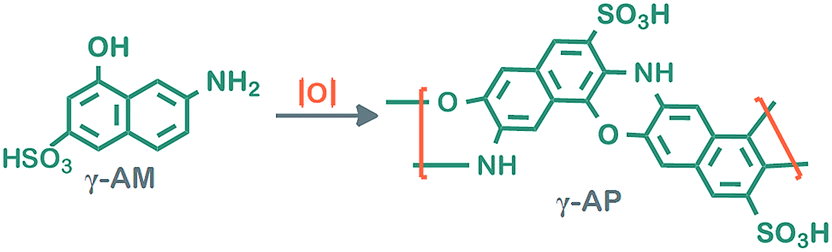 | ||
| Scheme 1 Synthetic pathway for γ-AP. | ||
Table 1 summarizes the conditions of oxidative polymerization for γ-AM with a 30% NaOCl solution in an aqueous alkaline medium. The solution color generally turned to a brownish color from pale yellow as soon as NaOCl was added to the reaction medium. During all reactions with high yields, the color of the solution changed slightly from yellow to black. The black-colored product is probably because of the hydroxylation reaction proceeding during polymerization.19 Accordingly, a highly efficient product is obtained very easily by OP. The conversion of γ-AM was 94% under optimum conditions ([M]0 = 0.025 mol L−1, [KOH]0 = 0.06 mol L−1, [NaOCl]0 = 0.08 mol L−1) at 80 °C for 15 h. The polymerization yield when using NaOCl as oxidant in aqueous basic medium was increased with an increase in temperature and then reduced over 90 °C due to the depolymerization of the polymer to γ-AM (see Table 1, runs 1–4). When the polymerization time at 80 °C was increased the maximum yield level was 85% for 15 h (runs 4–8). Finally optimum polymerization conditions were found by changing the initial concentrations of the alkaline and oxidant (runs 8–13). In these reactions, the maximum yield level of the polymer was significantly dependent on the temperature, initial concentrations of the alkaline and oxidant, and the time factor. The highest yield in OP using H2O2 as an oxidant was found to be 52% at 60 °C for 10 h. The yield level was decreased at all temperatures above 60 °C and times over 10 h. Accordingly, the optimum reaction conditions of γ-AP using H2O2 at 60 °C for 10 h were determined to be [M]0 = [KOH]0 = [H2O2]o = 0.08 mol L−1. Finally, the yield level of OP using air O2 was examined with regard to the different temperatures and times. The conversion of γ-AP was determined to be 68% at 60 °C for 5 h at the following concentrations: 2[M]0 = [KOH]0 = 0.08 mol L−1 and air = 8.5 Lsn−1. Under the same conditions, the conversion of γ-AP with air was less than that with H2O2 and NaOCl. As expected, the most suitable oxidant for the polymerization reaction was determined to be NaOCl.20 The synthetic route is presented in Scheme 1. Moreover, Scheme 1 reflects the formation of new 1,4-oxazine rings from a trifunctional monomer.
| Run | Temperature, °C | Time, s | [KOH]0, mol L−1 | [NaOCl]0, mol L−1 | Yield, % |
|---|---|---|---|---|---|
| a [M]0 = Monomer concentration, 0.025 mol L−1. | |||||
| 1 | 60 | 3 | 0.08 | 0.08 | 40 |
| 2 | 70 | 3 | 0.08 | 0.08 | 47 |
| 3 | 80 | 3 | 0.08 | 0.08 | 61 |
| 4 | 90 | 3 | 0.08 | 0.08 | 57 |
| 5 | 80 | 5 | 0.08 | 0.08 | 64 |
| 6 | 80 | 10 | 0.08 | 0.08 | 77 |
| 7 | 80 | 15 | 0.08 | 0.08 | 85 |
| 8 | 80 | 20 | 0.08 | 0.08 | 76 |
| 9 | 80 | 15 | 0.15 | 0.08 | 81 |
| 10 | 80 | 15 | 0.06 | 0.08 | 94 |
| 11 | 80 | 15 | 0.03 | 0.08 | 71 |
| 12 | 80 | 15 | 0.06 | 0.1 | 84 |
| 13 | 80 | 15 | 0.06 | 0.06 | 78 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1614, 1587 (Ar), 1307 (C–N), 1381 (OH), 1218 (C–O), 1162, 1033 (SO3), 815, 738 cm−1 (CH for substituted naphthalene ring). 1H-NMR (DMSO-d6): δ ppm, 10.63 (s, Hh, 1H), 7.32 (s, Hb, 1H), 7.20 (s, Hc, 1H), 7.07 (s, Hd, 1H). 13C-NMR (DMSO-d6): δ ppm, 149.87 (C4), 144.62 (C7), 139.67 (C6), 130.47 (C2), 127.14 (C8), 126.12 (C10), 124.69 (C9), 114.69 (C3), 114.86 (C1), 110.49 (C5).
N), 1614, 1587 (Ar), 1307 (C–N), 1381 (OH), 1218 (C–O), 1162, 1033 (SO3), 815, 738 cm−1 (CH for substituted naphthalene ring). 1H-NMR (DMSO-d6): δ ppm, 10.63 (s, Hh, 1H), 7.32 (s, Hb, 1H), 7.20 (s, Hc, 1H), 7.07 (s, Hd, 1H). 13C-NMR (DMSO-d6): δ ppm, 149.87 (C4), 144.62 (C7), 139.67 (C6), 130.47 (C2), 127.14 (C8), 126.12 (C10), 124.69 (C9), 114.69 (C3), 114.86 (C1), 110.49 (C5).Measurements
The infrared and ultraviolet-visible spectra were recorded on a PerkinElmer Spectrum One FT-IR using the system universal ATR sampling accessory within the wavenumbers of 4000–650 cm−1 and Analytikjena Specord 210 Plus, respectively. γ-AP was characterized by using 1H-NMR and 13C-NMR spectra (Bruker AC FT-NMR spectrometer operating at 400 and 100.6 MHz, respectively) at 25 °C by using deuterated DMSO as solvent at 25 °C. The surface morphology of the polymer was monitored by using a Jeol JSM-7100F Schottky field emission scanning electron microscope. Jeol 1400 plus model transmission electron microscopy (TEM) was used to determine the particle images of polymer. The samples were suspended in methanol and ultrasonically dispersed. Then 2 mL of this suspension was dried on a metal discs with carbon tape and carbon film 300 mesh Cu for SEM and TEM analyses, respectively. Topography and 3D images of the polymeric films were determined using atomic force microscopy (AFM) Alpha 300 A (WITec, Ulm, Germany). The samples were thermally annealed for 2 hours at 40 °C under vacuum before measurements. Specified surface areas of polymers were scanned angularly by cantilever of non-contact mod (AC, 42 N m−1, 285 kHz). X-ray diffractograms were obtained by using a PANalytical empyrean model X-ray diffractometer instrument with CuKα radiation at a wavelength of 1.54 Å over a 2θ range from 5° to 90°; the scan speed was 4° min−1. The number average molecular weight (Mn), weight average molecular weight (Mw) and polydispersity index (PDI) were determined by Gel Permeation Chromatography-Light Scattering (GPC-LS) device by Malvern Viscotek GPC Dual 270 max. For GPC investigations a medium 300 × 8.00 mm dual column addition 1 g L−1 of lithium bromide in DMF (1 mL min−1) was used as solvent. A light scattering detector (LS) and a refractive index detector (RID) were used to analyze the products at 55 °C. Touch equality was used to calculate the optical band gap (Eopt) of γ-AP. The CV measurements were carried out by a CHI 660 C electrochemical analyzer device (CH Instruments, Texas, USA). All the measurements were performed within a voltammetry cell containing an acetonitrile/DMSO solution (v/v, 1/4) under argon inert atmosphere at room temperature. The electrochemical potential of the silver electrode was calibrated according to the couple ferrocene/ferrocenium (Fc/Fc+). PL measurements were recorded in Hellma quartz cuvettes by a Shimadzu RF-5301PC spectrofluorophotometer. DSC analyses were performed by using a PerkinElmer Pyris Sapphire DSC instrument between 25–420 °C under nitrogen at a heating rate of 10 °C min−1 in an aluminum pan (in N2, 10 °C min−1). The TG thermograms were obtained with TG–DTA PerkinElmer Diamond system apparatus under the heating rates of 5, 10, 15, and 20 °C from ambient temperature up to 1000 °C. The experiments were carried out with polymer samples of 8–10 mg weight in dynamic nitrogen atmosphere at a flow rate of 60 mL min−1. A platinum crucible was used as a sample container. DMA tests were carried out using a PerkinElmer Pyris Diamond DMA 115 V. Rectangular samples of 10 mm (length) × 5.5 mm (width) × 0.6 mm (thickness) were used to conduct DMA tests in single cantilever bending mode at a frequency of 1 Hz, a heating rate of 3 °C min−1 and in the range 20–350 °C. The sample was prepared as follows: 0.5 g of polymer was placed in the titanium clamp (supplied from Triton Technology Ltd., United Kingdom) and extended followed by closing of the clamp from both sides by clamping.Results and discussion
The structure characterization of the polymer
γ-AP was prepared from γ-AM by oxidative polymerization under optimum reaction conditions using NaOCl. The yield of γ-AP is 94% after 15 h at 80 °C. The yields of other polymers are 68% using air and 52% using H2O2. These yields are much less compared to the yield obtained using NaOCl. γ-AP was obtained as dark black color powder. However, γ-AP exhibited higher solubility than that of the other phenol polymers in organic solvents.21 The tests were performed by using polymer/solution system (1 mg/1 mL) at room temperature. γ-AP was completely soluble in common organic solvents such as DMF, THF, DMSO, aqueous alkaline, and concentrated H2SO4, but it was partly soluble in toluene, benzene, acetone, acetonitrile, and ethyl acetate and insoluble in aprotic organic solvents including benzene, toluene and xylene.Fig. 1 represents the 1H-NMR spectra of γ-AM and γ-AP. Accordingly the γ-AM exhibited five signals in the aromatic region. The resonance signal positioned at 10.53 was due to the sulfonyl groups.22,23 The signals between 8.03–7.25 ppm were due to aromatic protons of the naphthalene ring. The broad but weak proton line in the range of 10.3–8.5 ppm was ascribed to the –OH proton resonance in the aminonaphthol ring because of the presence of highly intra- or inter-molecular hydrogen-bonded protons. This signal centered at 7.08 ppm was related to the NH2 protons of γ-AM. However, the 1H-NMR spectrum of the polymer presented a simple pattern with three signals of equal intensity. These signals centered at 7.32, 7.20 and 7.07 ppm were related to the Hb, Hc and Hd protons of γ-AP, respectively. The resonance signals He (7.39 ppm) and Hf (7.25 ppm) of γ-AM, disappeared in spectrum after polymerization compared to those of γ-AP, indicating that the polymerization occurred with the elimination of He and Hf protons from the naphthalene ring. The peaks posited at 10.63 and 8.14–7.65 ppm with lower proton intensity were assigned to the sulfonyl and terminal aromatic NH2 and NH3+ groups, respectively.24–26 There is also a broad peak centered at 3.86 ppm as a result of DMSO-d6 in the 1H-NMR spectra of γ-AP, similar to that of γ-AM. However, the absence of hydroxyl protons in the spectrum reflects the oxynaphthalene linkages in main chain.7 The aromatic proton signals also shifted to higher fields up to about 0.4–0.5 ppm after polymerization, indicating distributed conjugation due to the presence of etheric linking in the main chains.27,28 As a result, 1H-NMR measurements suggest that γ-AP was formed primarily through dehydrocoupling at the Hc and Hf position of the naphthalene ring.
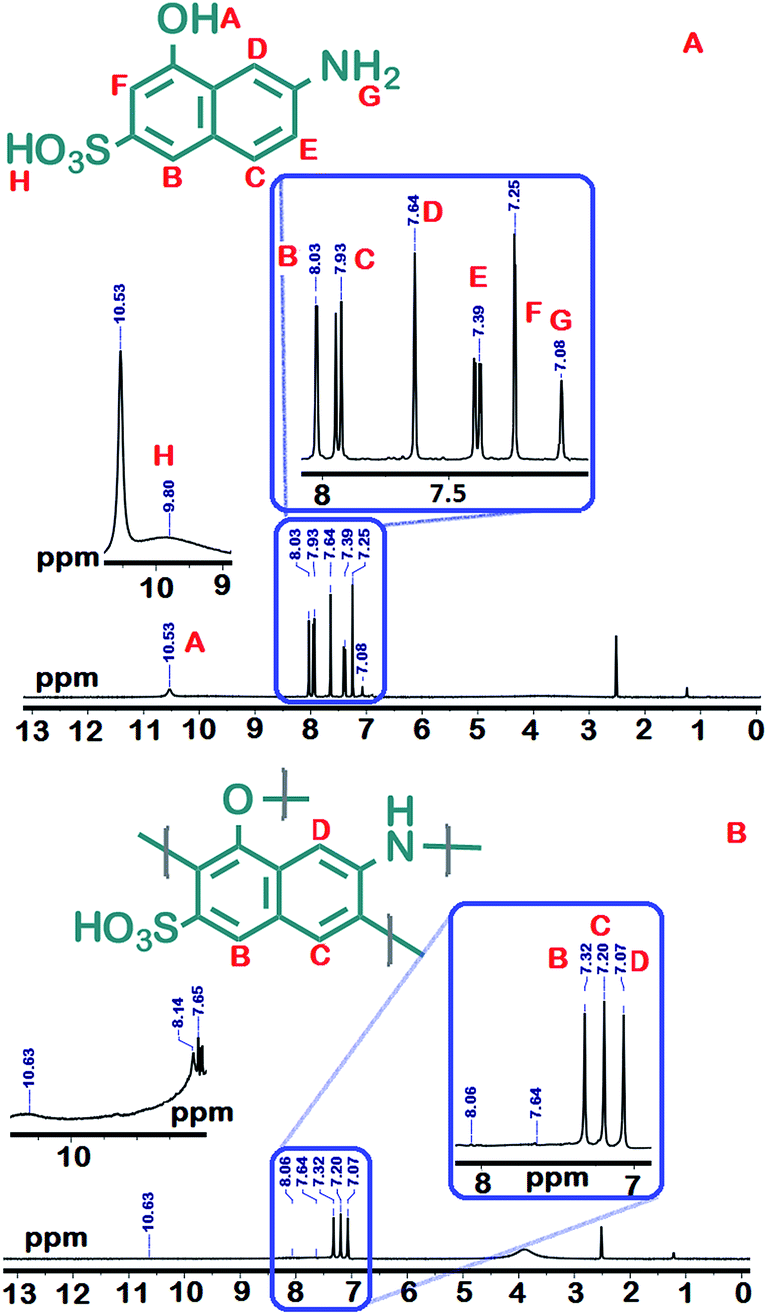 | ||
| Fig. 1 1H-NMR spectra of γ-AM (A) and γ-AP (B). | ||
Fig. 2 depicts the 13C-NMR spectra of γ-AM and γ-AP. Accordingly, γ-AP presented a total of nine carbon signals like γ-AM. The absence of extra carbon signals in the spectrum may be attributed to the non-branching polymer structure. The carbon signals C3 and C7 belonging to coupling centers of γ-AM were shifted to higher fields in the polymer spectrum, as expected. The other carbon signals are positioned with respect to coupling centers. These findings confirmed that polymerization occurred with high regioselectivity.
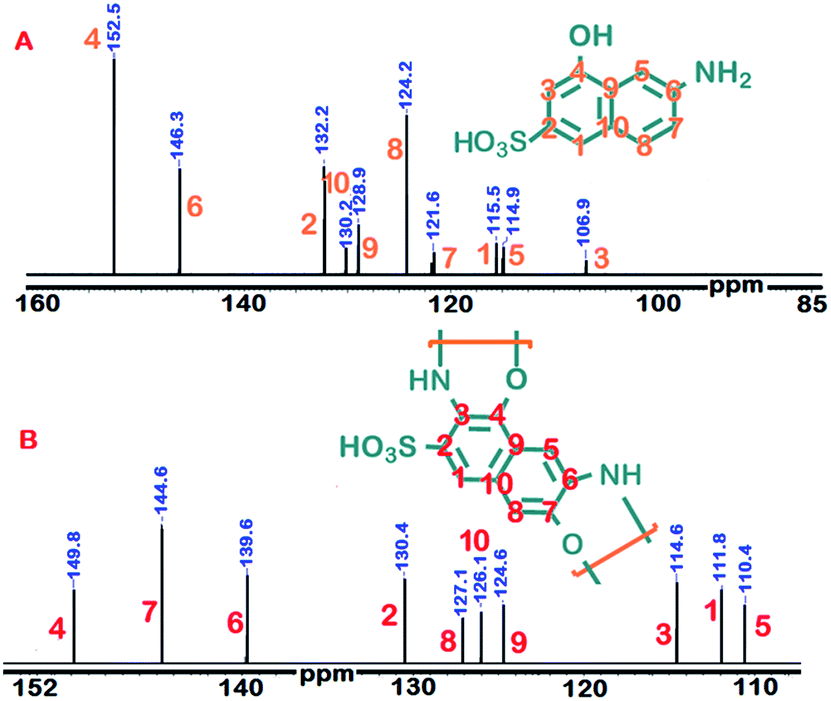 | ||
| Fig. 2 13C-NMR spectra of γ-AM (A) and γ-AP (B). | ||
The polymerization of the phenolic and aniline-based monomers with an oxidant in alkaline medium occurs via a radicalic mechanism.29 For polymerization of γ-AM, two radicalic combinations, which are different from each other because of the bifunctional groups, are suggested. The possible radicalic combinations of γ-AM are exhibited in Scheme 2. Positions R1, R2, R3, R4, RA, RB and RF on the naphthalene ring were determined to be conceivable coupling modes for polymerization. However, the most probable coupling modes for polymerization were the R2 and RB positions because they were positioned very close to the electronegative groups. Therefore, a highest propensity for bond formation was probable at R2 and RB positions.30
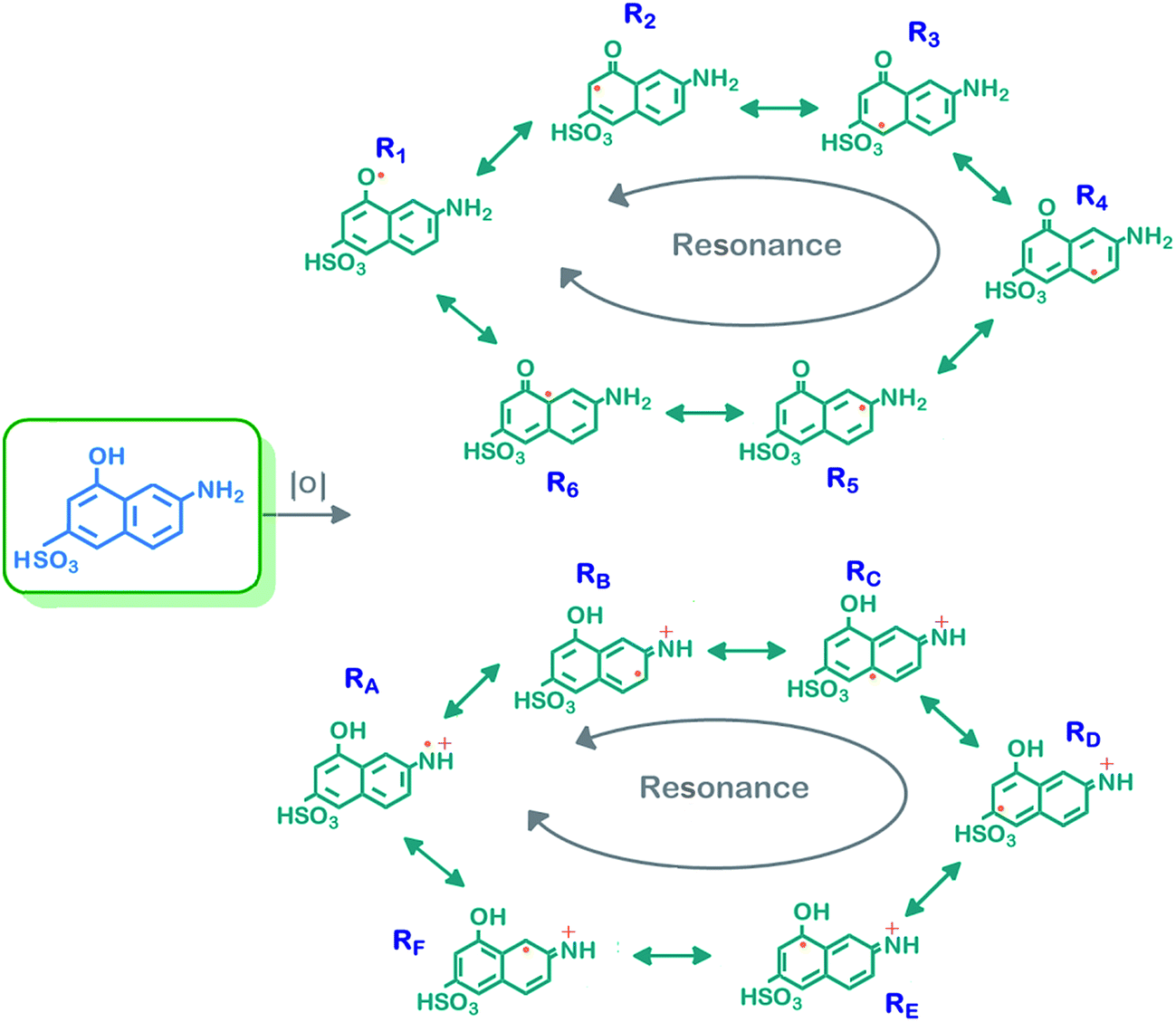 | ||
| Scheme 2 Possible resonance forms of γ-AM. | ||
UV-Vis spectrum of γ-AP is given in Fig. 3(a). The absorption spectrum of γ-AP has a maximum absorption wavelength (λmax) of 374 nm compared to γ-AM. The spectrum exhibited a red shifted absorption maximum with a tail from 340 to 700 nm due to the π-conjugated structure. An extended absorption spectrum generally indicates the coexistence of both long and short effective conjugation in the main chain and an increase in the molecular weight after polymerization. The optical band gap of γ-AP can be determined by touch equality as the classical relation in semiconductor.31
| αhv = A(hv − Eg)n | (1) |
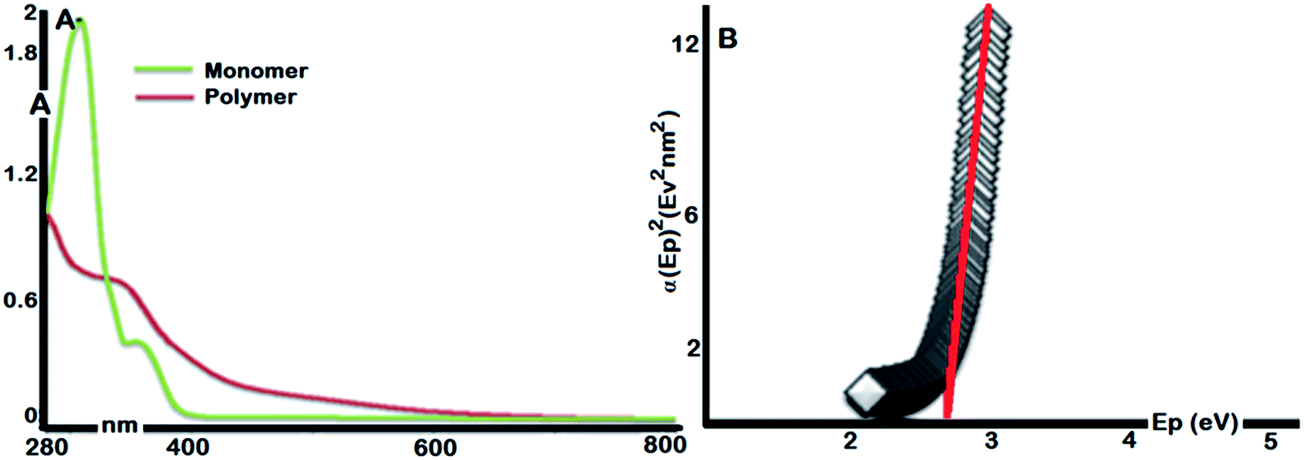 | ||
| Fig. 3 UV-Vis spectra of γ-AM and γ-AP (A) and Touch plot for γ-AP (B). | ||
The representative FTIR spectra of γ-AM and γ-AP are given in Fig. 4. As seen in Scheme 1, γ-AM has three functional groups and also contains many coupling sites for polymerization. However, IR and NMR studies provide some important information about the chemical structure of γ-AP. The FT-IR spectrum of γ-AP, compared to γ-AM, showed more shallow and broad peaks because of π-conjugation in the polymer chain. The peak broadenings and shifts in the spectrum were attributed to the polymer formation. The vibration bands observed at approximately 3323 and 3187 cm−1 were due to the characteristic O–H and N–H stretching of γ-AM, respectively. The intensity of these vibrations drastically increased after polymerization. So-called bands were also shifted to a longer wavenumber, which was ascribed to intramolecular or intermolecular hydrogen bonds originating from self-doping of the sulfonyl groups to the proton acceptors like –NH groups in conjugation.32,33 However, the spectra displayed no specific bands in the region at nearly 1700 cm−1, which corresponded to the oxidation peak of hydroxyl groups. The strong bands centered at 1643 cm−1 and its shoulders at 1614 and 1587 cm−1 were related to the CC stretching vibrations of the conjugated structure.10 The broad band at 1405 cm−1 in the spectrum of the polymeric product was assigned to C–C stretching vibrations in the benzenoid ring. A dominant band observed at 1307 cm−1 was also attributable to C–N stretching in the main chain. The bands at 1162 and 1033 cm−1 in the polymer spectrum were related to the asymmetric and symmetric stretching vibrations of sulfonyl groups, respectively. Also the two new bands at 815 and 738 cm−1 reflected the aromatic C–H in-plane deformation vibrations of the trisubstituted naphthalene ring. In the spectrum, the presence of aromatic C–O–C couplings located at 1218 cm−1 and absence of phenolic OH in-plane bending vibration in the region 1320–1370 cm−1, as well as the CO stretching vibrations (Pumerer's ketone structure) at nearly 1700 cm−1, indicated that all of the hydroxyl groups take part in coupling reactions during the polymerization process.
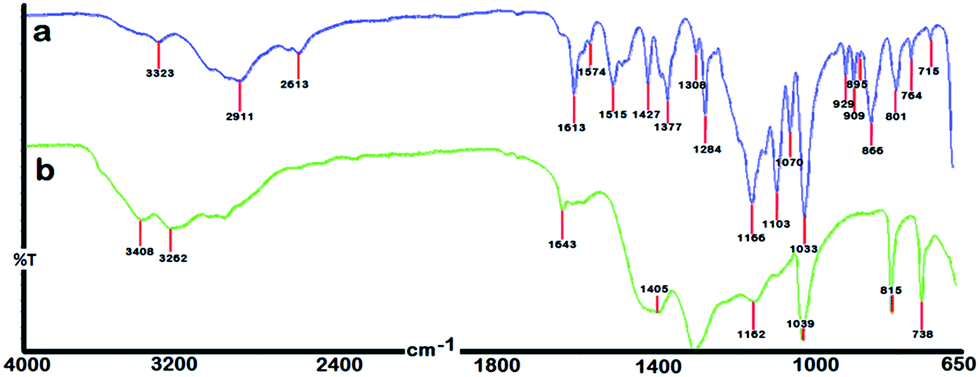 | ||
| Fig. 4 FT-IR spectra of γ-AM (a) and γ-AP (b). | ||
SEC analyses showed Mn and PDI values of γ-AP were 32000 Da and 1.42, respectively. Accordingly, higher solubility of γ-AP could be attributed to its relatively low average molecular weight. The thermal stability is a measure of the availability of materials in industry. In order to determine the thermal stability and kinetic parameters related to the solid state decomposition of a material, the thermogravimetry technique is generally used. Here, the thermal stability and kinetic parameters of γ-AP particles were determined by TG–DTG–DTA and DSC instruments. DSC measurements were carried out at a heating rate of 10 °C min−1 ranging from 25 to 420 °C in N2. The TG–DTG–DTA analysis of γ-AP was performed by heating up to 1000 °C at heating rates of 5, 10, 15 and 20 °C min−1. All measurements were repeated until reproducible results were obtained. The TG–DTG–DTA curves of γ-AM and γ-AP are given in Fig. 5. The initial and final temperatures of decomposition, losses of mass in % and the temperatures related to the maximum decomposition rate were determined from TG–DTG–DTA curves.
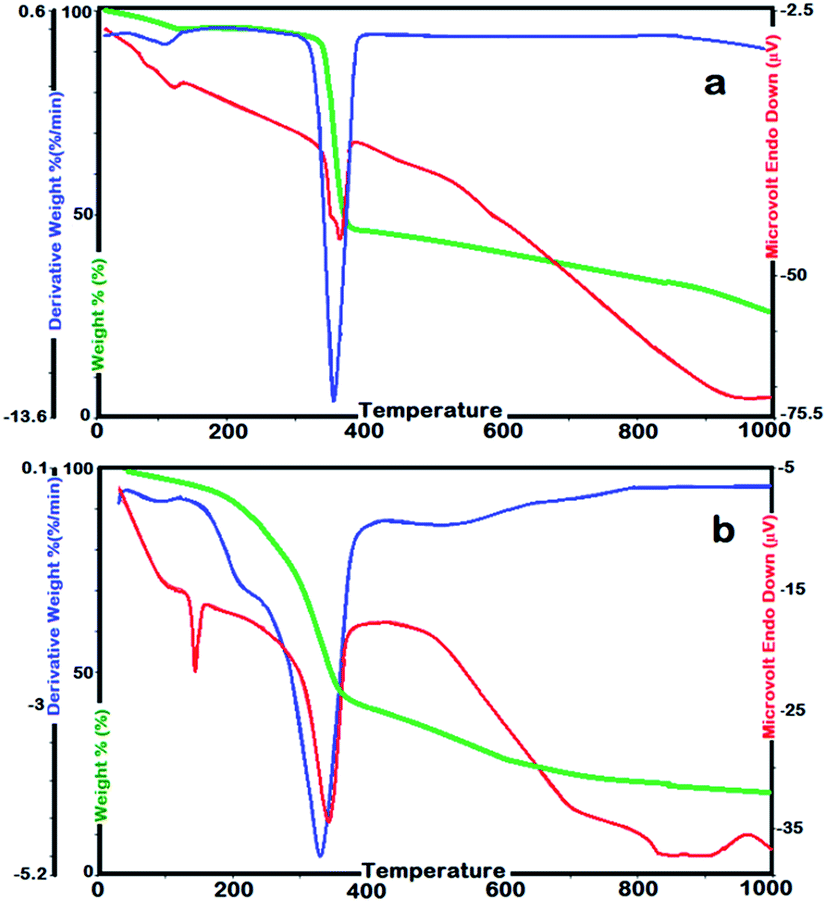 | ||
| Fig. 5 Typical TG, DTG and DTA curves for γ-AM (a) and γ-AP (b) at heating rate of 10 °C min−1. | ||
Accordingly, on the TG curve (Fig. 5(a)), γ-AM presented one step decomposition over the temperature range 362–389 °C with a mass loss 72.1% in weight. The endothermic thermal effect at 367 °C in the DTA profile was related to the decomposition step of γ-AM. γ-AP decomposed in one stage over the temperature range 210–383 °C with a mass loss of 90.2%. Initial temperature of decomposition and amount of waste carbon of γ-AP was lower than those of γ-AM due to the oxynaphthalene linkages in main chain. The corresponding DTA profile displayed two endothermic peaks. The first endothermic peak observed at 121 °C was attributed to the glass transition temperature of nanofiller γ-AP. The second one at 374 °C was related to the decomposition stage of γ-AP. The temperature values related to the maximum decomposition rate for γ-AM and γ-AP were determined as 348 °C and 361 °C, respectively.
Also, the DSC curve of γ-AP presented three different regions physically separated from each other (Fig. 6(a)). The endothermic effects at 42 and 78 °C belong to the evaporation of water absorbed by γ-AP. The endothermic peaks observed at 176 and 373 °C illustrate the melting and decomposition temperatures of γ-AP. The DSC curve also presented multiple peaks due to ionic cross-links in the glass transition region. The ionic cross-links contribute a relaxation of the barrier to bond rotation around the polymer backbone and intermolecular constraints. The volume shrinkage caused by these relaxations appears as a drop in temperature in DSC thermograms.34 Here the main chain contains sulfonyl groups showing ionic effects and cross-linking causing oxynapthalene units. However, a DMA analysis was carried out to identify the glass transition temperature of γ-AP precisely (Fig. 6(b)). The type of DMA curves represented the semiconductive nature of conjugated polymer. The high slope of the tanδ curve is due to cross-linking in the polymer structure. The glass transition and melting temperature of the resulting polymer were clearly observed at 123 and 174 °C, respectively.
 | ||
| Fig. 6 DSC (A) and DMA (B) curves of γ-AP. | ||
On the other hand, in order to calculate the kinetic parameters related to the solid state decomposition of γ-AP, several methods based on multiple heating rates were used. TG–DTG–DTA thermograms of γ-AP were recorded in dynamic N2 at the heating rates of 5, 10, 15, and 20 °C min−1. All thermograms indicated that the solid state decomposition reaction takes place in one step during the heating program. As expected, the initial temperature of decomposition shifted to higher values with an increase in heating rate and also the amount of carbon residue increased as a result of decomposition. All thermograms obtained had similar characteristics. In the TG–DTG curves of γ-AP, no water loss was observed. The temperature values related to the maximum decomposition rate from DTG curves was found to be 355.7, 367.9, 372.0 and 381.4 °C, respectively, for the heating rates of 5, 10, 15 and 20 °C min−1. In many studies, isoconversional methods, based on multiple heating programs, were reported as the most reliable methods to determine the kinetic parameters of thermally activated reactions.35 In the present work, the kinetic parameters related to the thermal decomposition of γ-AP were calculated by different isoconversional methods. The kinetic parameters related to the solid state decomposition of γ-AP were determined using Kissinger36 and Kim-Park methods,37 without precise knowledge of the reaction mechanism, as described earlier. They used the temperature values corresponding to the maximum rate of solid state decomposition. The linear least squares fit for both isoconversional methods in the range of 0.05 < α< 0.95 gave good values of regression coefficient. The activation energy values were evaluated as 179.7 and 190.6 kJ mol−1, respectively, for Kissinger and Kim-Park methods from the slope of the linear least squares plot. Besides these methods, isoconversional integral methods like Tang,38 KAS,39 FWO,40 Starink,41 and Bosewell,42 and an isoconversional differential method like Friedman,43 were used to calculate the kinetic parameters related to the solid-state thermal decomposition of γ-AP. The invariant activation energies were calculated by the conventional integral procedures such as FWO, KAS, Tang, Starink and Bosewell though in the case of variable activation energies they might provide incorrect values. In those cases, advanced integral isoconversional or differential methods are applied instead of integral isoconversional methods. Thus, in the present case, we used both differential isoconversional methods (Friedman and Kissenger) and integral isoconversional methods (FWO, KAS, Tang, Starink and Bosewell) for solid state decomposition kinetic studies. The average values of the activation energies calculated by using the methods of Tang, KAS, FWO, Starink, Bosewell and Friedman were determined to be 184.4, 184.4, 185.4, 188.5, 183.6 and 187.1 kJ mol−1, respectively. The activation energies, very close to together, show that the kinetic analysis was successfully performed by different isoconversional methods. The average values of activation energy obtained by using different isoconversional methods for the solid stage thermal decomposition stage of γ-AP, over a wide range of conversions, are also given in Table 2.
| Conversions degree | ETang | EKAS | EFWO | EStarink | EBosewell | EFR |
|---|---|---|---|---|---|---|
| 0.05 | 173.7 | 175.8 | 172.9 | 177.6 | 173.4 | 176.4 |
| 0.1 | 185.0 | 184.3 | 185.6 | 187.8 | 182.3 | 186.2 |
| 0.2 | 186.7 | 185.9 | 186.4 | 188.2 | 183.4 | 188.2 |
| 0.3 | 187.8 | 187.2 | 186.5 | 189.5 | 185.6 | 188.9 |
| 0.4 | 189.8 | 189.3 | 189.1 | 192.3 | 187.9 | 191.6 |
| 0.5 | 187.5 | 187.0 | 186.7 | 189.4 | 185.6 | 188.8 |
| 0.6 | 184.5 | 184.1 | 184.2 | 187.2 | 182.7 | 186.1 |
| 0.7 | 183.1 | 182.6 | 184.0 | 187.7 | 181.5 | 185.3 |
| 0.8 | 188.1 | 187.6 | 188.3 | 191.8 | 185.4 | 189.2 |
| 0.9 | 190.9 | 190.3 | 190.9 | 193.6 | 188.9 | 191.1 |
| 0.95 | 177.9 | 174.9 | 180.4 | 183.6 | 172.8 | 181.2 |
| Average | 184.4 | 184.4 | 185.4 | 188.5 | 183.6 | 187.1 |
Also, the correlation coefficients of the Arrhenius type plots obtained by all the methods are considerably higher in the region of α = 0.05–0.95 in N2. Table 2 also presents the dependence of the conversion degree on activation energy in N2. The E-dependencies degree of conversion for the solid state thermal decomposition stage in the thermal decomposition process of γ-AP had a similar behavior for all the methods with conversions ranging from α = 0.05 to 0.95. This indicates that the mechanism related to the thermal process of γ-AP does not change with the conversion degree 0.05 < α < 0.95. The lowest activation energy value related to the initial decomposition of γ-AP was determined to be approximately 172.9 kJ mol−1 by using the FWO method at progressive conversion values. In the last part of this study, the master plot curves were used to determine the most likely mechanism corresponding to the solid state thermal decomposition process of γ-AP. The kinetic model related to the thermal decomposition stage of γ-AP was found by a comparison of the experimental master plots with theoretical ones corresponding to the mechanism of a solid state decomposition process. The comparisons indicate that the kinetic process agreed with the D6 master curve very well. Thus the master plot curves suggest a diffusion-type kinetic model for the solid state thermal decomposition stage of γ-AP. As reported in the literature, D6 model suggests a three-dimensional counter diffusion type decomposition mechanism and is also known as Komatsu–Uemura or anti-Jander equation.44
Electrochemical properties
CV measurements are widely used as an effective method in which data is provided about semiconductor properties. Some physical parameters like electrochemical band gap can be determined from a single measurement. Fig. 7(a) depicts the cyclic voltammograms of γ-AP at various scan rates ranging from 50 to 600 mV s−1. All the measurements were carried out by using a triple electrode system. In the measurements, GCA was used as working electrode, Ag wire as reference electrode and Pt wire as auxiliary electrode. Voltammograms of γ-AP were obtained in a mixture of ACN/DMSO (1/4, v/v) solvent containing 0.1 M tetrabutylamonium hexafluorophosphate as a supporting electrolyte. In order to investigate its electroactive nature, the scanning rate was chosen as 50, 100, 200, 300, 400, 500 and 600 mV s−1. Electrochemically the band gap of γ-AP was calculated by the equations used in a previous study.45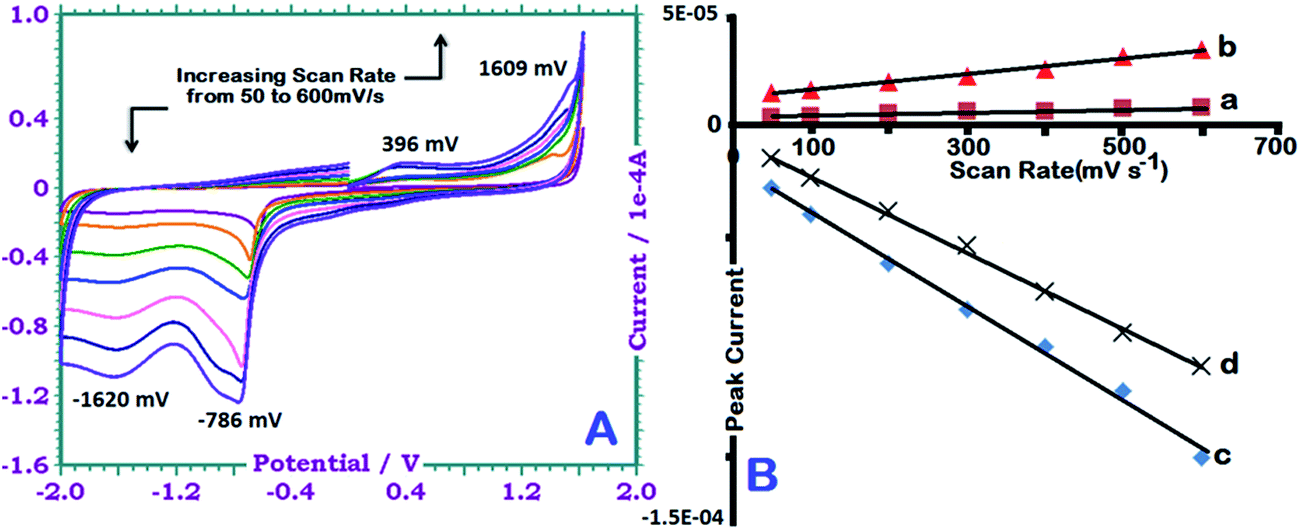 | ||
| Fig. 7 CVs of γ-AP in ACN/DMSO (v/v, 4/1) at different scan rates (from bottom to top at oxidation area): 50, 100, 200, 300, 400, 500 and 600 mV s−1 (A). Polymer conc: 0.1 g L−1. The peak currents vs. scan rates at +396 (a), +1609 (b), −786 (c) and −1620 (d) mV (B). | ||
In the anodic region γ-AP indicated two particular peaks at 396 and 1609 mV. The first peak at 396 mV was attributed the oxidation of intermolecular –NH– and/or free (unlinked) –NH2 groups to form a polaron structure.46 The next peak was related to the oxidation of free –OH groups (Pummerer's ketone).47 The reduction peaks at −1620 and −786 mV in the cathodic region were due to the neutralization of the oxidized state of the polymer and the reduction of ketone groups to hydroxyl groups, respectively. Fig. 7(b) presents the relationships of current versus scanning rate. Also Fig. 7(b) clearly illustrate that the anodic and cathodic peak currents increase linearly with increasing scan rate. This indicates a diffusion-controlled redox process.48 The regression coefficients of straight lines was greater than 0.98. However, for γ-AP, the energy levels of EHOMO–ELUMO were calculated as −5.99 and −3.19 eV respectively; the electrochemical band gap was found to be approximately 2.81 eV. This value is slightly higher than the optical band gap due to an interfacial barrier between the polymer film and the electrode surface.49
Surface properties
X-ray diffraction patterns for γ-AM and γ-AP particles are presented in Fig. 8. XRD analyses of the powdered γ-AP particles were carried out at 2θ angles in the range of 0–70°. The γ-AM exhibited many sharp and strong peaks at low Bragg angles, which reveal that γ-AM is a crystalline monomer. The diffraction peaks of γ-AP particles were at almost the same positions as those of γ-AM in the range of 10–30°, showing that γ-AP particles consisted of repetitive units.50 The peak intensities of γ-AP particles at low Bragg angles were lower than those at 2θ = 20–35° because it does not contain many more frees OH/NH2 groups. This indicates that there were smaller crystalline particles within the polymer.51 The first peaks at 2θ = 11.24° and 16.9° (corresponding to d-spacing of 3.9 Å) and the second peak at 2θ = 16.9° (corresponding to d-spacing of 2.7 Å) were attributed to parallel repeating units of γ-AP.52 The peaks in the range of 20–25° may be caused by the periodicity perpendicular to the semicrystalline main chains.53 In addition, the peak at 2θ = 27.7° was assigned to a periodicity caused by H-bonding between γ-AP chains.54 As a result, γ-AP particles presented a heterogeneous system consisting of a partly crystalline region. The percentage crystallinity value of γ-AP was determined to be approximately 21.6%. These findings are also supported by SEM, AFM and especially TEM analyses.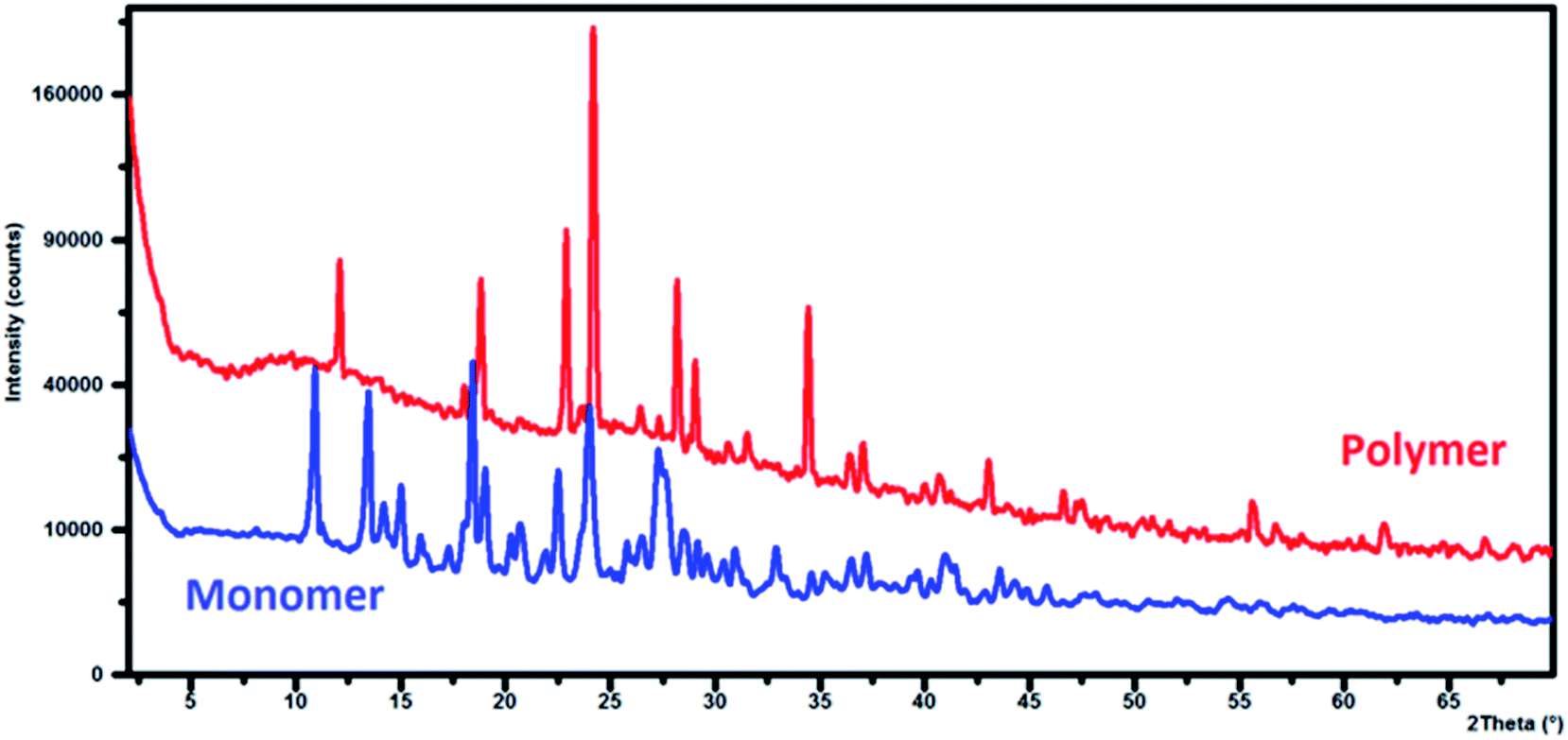 | ||
| Fig. 8 Te XRD patterns of monomer (γ-AM) and polymer (γ-AP). | ||
The survey morphology of the γ-AP particles was investigated by a scanning electron microscope (SEM) (FEG SEM Philips XL-305 instrument). SEM images are shown in Fig. 9. Accordingly, the images presented an inhomogeneous and random dispersion, while also exhibiting fine channels and agglomerates on the surface. The agglomerations reveal a crystal lattice structure consisting of linear forms with approximately 1 μm in size.
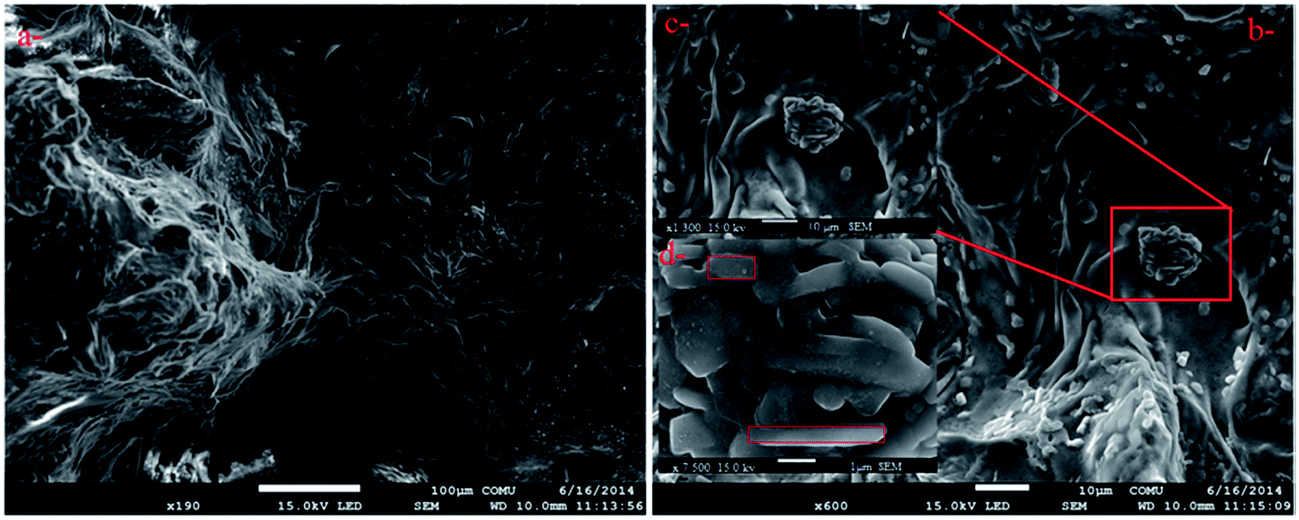 | ||
| Fig. 9 SEM photographs of γ-AP particles with different magnifications. ((a) bar shows 100 μm, (b) and (c) bar shows 10 μm, (d) bar shows 1 μm, respectively.) | ||
The topography of the polished film surface of γ-AP was studied by using an AFM image. The resulting topography is illustrated in Fig. 10. The topographies are presented in three dimensions as originally recorded. They exhibited a typical nodular structure for γ-AP. The RMS roughness of the film was found to be 14.7 nm. The nodule size was nonhomogeneous and changed from 15 up to 400 nm in diameter. This may be attributed to the cross-linked network developed during polymerization. These topographies are in agreement with those of the SEM investigation.
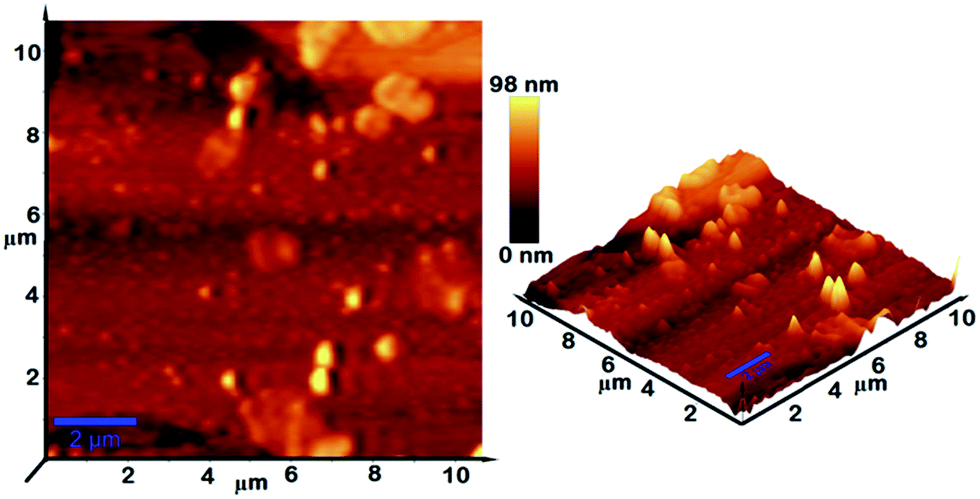 | ||
| Fig. 10 Three-dimensional AFM images of γ-AP film (3D flattened images). | ||
A Tecnai G2 Spirit Biotwin model high contrast transmission electron microscope (C-TEM) was used to investigate the nanomorphology of γ-AP particles. The samples were suspended in methanol and ultrasonically dispersed. TEM images of γ-AP are given in Fig. 11. The images clearly reveal the formation of 1D nanofiber structures. The average diameters of the resulting nanofibers were approximately 5–10 nm. The nanofiber length also ranges from 10 nm to 500 nm. Long and thin nanofiber structures were evident for the partially crystalline structure of γ-AP. TEM and XRD results supported each other, but TEM and SEM images seemed different. This difference was probably caused by different test conditions or heterogeneous polymer morphology. When a conjugated polymer is doped with a suitable doping agent, the morphology and interchain packing distance of the polymer are affected. In many studies the influence of various doping agents on morphology of the conjugated polymers was investigated.55 γ-AP is a self-doped conjugated polymer due to the sulfonyl groups. Here, without using any doping agent, we easily prepared a nanofiber structure by using a green process.
 | ||
| Fig. 11 TEM images of γ-AP particles ((a) bar shows 1 μm, (b) bar shows 2 μm). | ||
Conductivity properties
The conductivity of I2-doped and self-doped γ-AP was investigated by using the standard four-point probe technique. The powder sample was, made into pellets by compressing with 1600 kg cm−1 hydraulic pressure. Then the pellets were exposed to iodine vapors in a desiccator for up to 7 days. The self-doped conductivity of γ-AP was found to be 7.1 × 10−8 S cm−1. When treated with iodine vapor, its conductivity increased significantly and then remained constant after reaching a saturation level. The maximal conductivity for γ-AP was about 8.3 × 10−6 S cm−1 (for 120 h). An increase in conductivity is due to a charge-transfer complex formed by the interchain and intrachain electrostatic interactions between iodine molecules and naphthalene rings as well as free amino groups.56 Although conjugation was distributed by means of oxynaphthalene linkages, γ-AP had relatively higher conductivity than those of previously synthesized phenol polymers.57 As a result γ-AP particles can be an alternative gas sensor due to fast conductivity.Photophysical properties
The PL spectra of γ-AM and γ-AP A was obtained by a Shimadzu RF-5301PC spectrofluorophotometer. In the measurements, fluorescein solution in 0.1 M NaOH was used as a standard sample. The PL spectrum of γ-AM is shown in Fig. 12. The PL spectrum of γ-AP in DMSO solution was different to that of γ-AM in respect to some PL characteristics like the shape, position, and intensity. γ-AM emits a blue light with an emission maximum at 408 nm when excited at 350 nm in DMSO. In addition, γ-AM exhibited no red-shifting in emission with increasing excitation wavelengths and its emission was also quenched. However, under the same experimental conditions, γ-AP surprisingly displayed a different emission maximum at different excitation wavelengths.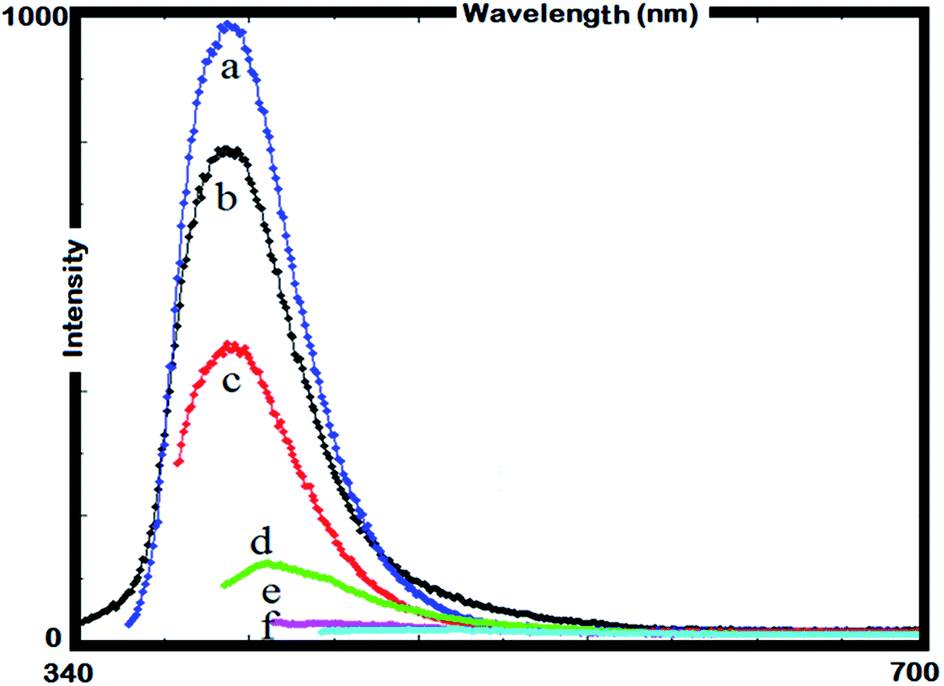 | ||
| Fig. 12 PL spectrum of γ-AM in DMSO, λEx: 330 (a), 340 (b), 350 (c), 360 (d), 370 (e), and 380 (f) (slit width: 5 nm and conc.: 10 mg L−1 in all measurements). | ||
Accordingly, γ-AP emits a multi-colour light including blue, yellow and red when excited at 380, 420 and 500 nm, respectively, which was attributed to its polyconjugated backbone. The emission colors are given in the photographs located in Fig. 13. The fluorescence quantum yields (Φf) of γ-AP solution were calculated from a comparative method suggested by Williams et al.58 and determined to be 1.4, 0.7 and 0.9% for blue, green, and red PL, respectively. The Φf of γ-AP drastically decreases with increasing excitation wavelengths.
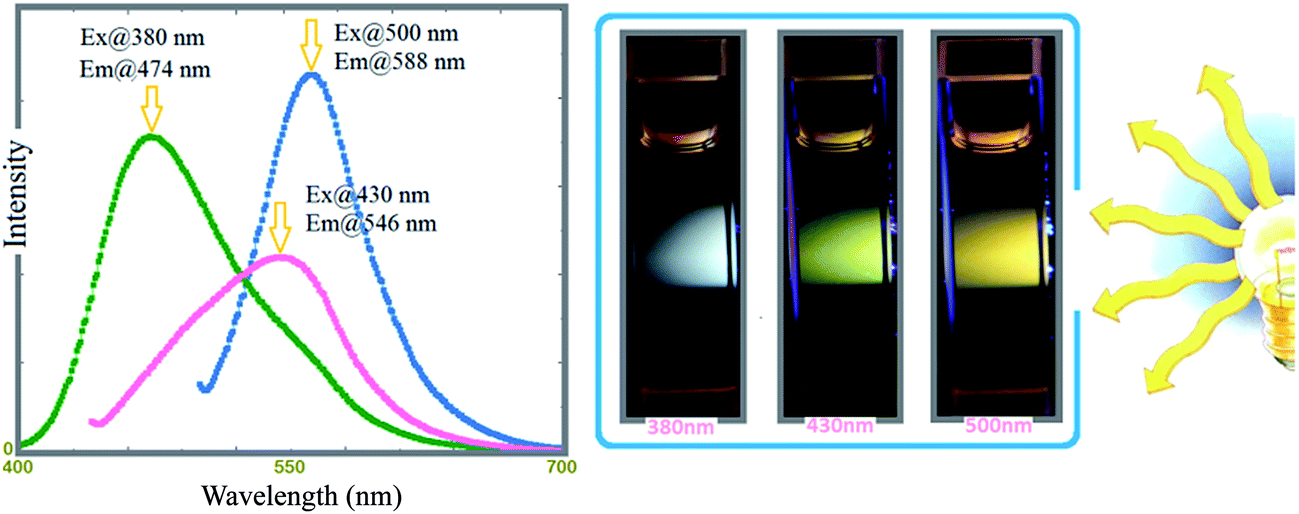 | ||
| Fig. 13 Normalized emission spectra of γ-AP for three excitation values; 380, 430, and 500 nm, respectively. Slit width: 5 nm and conc.: 10 mg L−1 in all measurements. | ||
To confirm the multicolor emission behavior of γ-AP by changing the excitation wavelength, confucal microscope analysis (Leica TCS SPE model) was also performed. The γ-AP solution of a given amount was first dropped on a glass sheet and then dried. The obtained sample was separately excited at 380, 430, and 500 nm to observe blue, green, and red emissions, respectively. The emissions colors agreed with the PL ones very-well (Fig. 14). The relatively pale emission colors are due to the low quantum yields of γ-AP. As a result, γ-AP exhibited an adjustable PL color by changing the source light energy on the desired scale. So far, several polymeric systems like oligopyrene nanofibers,59 polyvinylpyridine gels,60 polyamidoamine nanoparticles,61 hyperbranched polyamidoamines,62 nitro-substituted pyrene oligomers63 and oligo(4-hydroxyquinoline)64 have unusual multicolor emission behaviors. This phenomenon is associated with broad chain dispersity of the conjugated polymer considered to be a multi-chromophoric system. Each unit with different conjugation lengths behaves as a different chromophore group. Accordingly the light with longer wavelength is absorbed by the longer conjugated chains and vice versa.
 | ||
| Fig. 14 Confucal microscope images of γ-AP. The excitation wavelengths were 380 (a), 430 (b), and 500 nm (c). | ||
Conclusions
In conclusion, we report a novel regioselectively-functionalized polymer from a trifunctional monomer by a simple and environmentally friendly synthetic method. We also discovered some properties of γ-AP as given below. Accordingly, (i) optimum polymerization conditions for γ-AM were determined to be [M]0 = 0.025 mol L−1, [KOH]0 = 0.06 mol L−1, [NaOCl]0 = 0.08 mol L−1 at 80 °C for 15 h. Under these conditions the maximum yield level was 94%, (ii) the most suitable oxidant for the polymerization reaction under optimum conditions was NaOCl, (iii) spectral analyses indicated γ-AP consisted of regioselectively-functionalized 1–4 oxazine rings, (iv) γ-AP presented an average molecular weight of 32000 Da and polydispersity index of 1.42, (v) γ-AP has an optical band gap value of 2.68 eV for an indirect forbidden transition and also an electrochemical band gap value of 2.81 eV for a diffusion-controlled electrode process, (vi) γ-AP has adjustable PL color, (viii)XRD, DSC, DMA, SEM and TEM analyses suggested a semicrystalline-nanofiber structure for γ-AP, (ix) the conductivity of iodine doped γ-AP was found to be 8.3 × 10−6 S cm−1 for 120 h, (x) the average value of the activation energy related to the solid state thermal decomposition of γ-AP was calculated to be approximately 185 kJ mol−1 by using isoconversional methods, and (xi) finally, γ-AP exhibited a diffusion-type kinetic model for thermal decomposition mechanism. Thus, these results indicate γ-AP may be considered a promising polymer for some applications such as rechargeable batteries, light-emitting diodes, biosensors, and electrochromics in academia and industry.
Acknowledgements
The authors thank the Scientific and Technological Research Council of Turkey (Project Nu.: KBAG-113Z587) for financial support of this work.References
- Y. Gupta, K. Hellgardt and R. J. Wakeman, J. Membr. Sci., 2006, 282, 60–70 CrossRef CAS PubMed.
- S. Radhakrishnan and S. B. Kar, Sens. Actuators, A, 2005, 120, 474–481 CrossRef CAS PubMed.
- A. Bishop and P. Gouma, Rev. Adv. Mater. Sci., 2005, 10, 209–214 CAS.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314–315 CrossRef CAS PubMed.
- T. C. Girija and M. V. Sangaranarayanan, J. Power Sources, 2006, 159, 1519–1526 CrossRef CAS PubMed.
- F. Doğan, A. Bilici, M. Yıldırım and İ. Kaya, Sci. Adv. Mater., 2014, 6, 1957–1964 CrossRef PubMed.
- A. Bilici, F. Doğan, M. Yıldırım and İ. Kaya, React. Funct. Polym., 2011, 71, 675–683 CrossRef CAS PubMed.
- A. Bilici, F. Doğan and İ. Kaya, Ind. Eng. Chem. Res., 2014, 153, 104–109 CrossRef.
- F. Doğan, İ. Kaya and K. Temizkan, Eur. Polym. J., 2015, 66, 397–406 CrossRef PubMed.
- H. Bhandari, S. Singh, V. Choudhary and S. K. Dhawan, Polym. Adv. Technol., 2011, 22, 1319–1328 CAS.
- J. Kadota, T. Fukuoka, H. Uyama, K. Hasegawa and S. Kobayashi, Macromol. Rapid Commun., 2004, 25, 441–444 CrossRef CAS PubMed.
- M. Kurisawa, J. E. Chung, H. Uyama and S. Kobayashi, Macromol. Biosci., 2003, 3, 758–764 CrossRef CAS PubMed.
- R. Chelikani, Y. H. Kim, D. Y. Yoon and D. S. Kim, Appl. Biochem. Biotechnol., 2009, 157, 263–277 CrossRef CAS PubMed.
- J. K. Stille, Macromolecules, 1981, 14, 867–870 CrossRef.
- G. Jegou and S. A. Jenekhe, Macromolecules, 2001, 34, 7926–7928 CrossRef CAS.
- A. Bilici, İ. Kaya and F. Doğan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2977–2984 CrossRef CAS PubMed.
- A. Bilici, F. Doğan, M. Yıldırım and İ. Kaya, Mater. Chem. Phys., 2013, 140, 66–74 CrossRef CAS PubMed.
- F. Doğan, İ. Kaya and K. Temizkan, J. Macromol. Sci., Part A: Pure Appl.Chem., 2014, 51, 948–961 CrossRef PubMed.
- T. Oguchi, S. Tawaki, H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1999, 20, 401–403 CrossRef CAS.
- İ. Kaya, F. Doğan and A. Bilici, Polym. Int., 2009, 58, 570–578 CrossRef PubMed.
- A. Bilici, İ. Kaya and M. Sacak, J. Inorg. Organomet. Polym., 2010, 20, 124–133 CrossRef CAS PubMed.
- E. Pretsch, P. Bühlmann and C. Affolter, Structure Determination of Organic Compounds Tables of Spectral Data, Springer-Verlag, Berlin, 2000 Search PubMed.
- S. Mu, J. Phys. Chem. B, 2008, 112, 6344–6349 CrossRef CAS PubMed.
- A. M. Catargiu and M. Grigoras, Acta Chem. Iasi, 2011, 19, 81–100 CAS.
- W. C. Chen and S. A. Jenekhe, Macromolecules, 1992, 25, 5919–5926 CrossRef CAS.
- M. R. Huang, X. G. Li, Y. L. Yang, X. S. Wang and D. Yan, J. Appl. Polym. Sci., 2001, 81, 1838–1847 CrossRef CAS PubMed.
- J. Xu, Y. Zhang, J. Hou, Z. Wei, S. Pu, J. Zhao and Y. Du, Eur. Polym. J., 2006, 42, 1154–1163 CrossRef CAS PubMed.
- M. Lanzi and L. Paganin, React. Funct. Polym., 2010, 70, 346–360 CrossRef CAS PubMed.
- H. Mart, Des. Monomers Polym., 2006, 9, 551–588 CrossRef CAS PubMed.
- B. Lu, J. Yan, J. Xu, S. Zhou and X. Hu, Macromolecules, 2010, 43, 4599–4608 CrossRef CAS.
- K. Colladet, M. Nicolas, L. Goris, L. Lutsen and D. Vanderzande, Thin Solid Films, 2004, 451–452, 7–11 CrossRef CAS PubMed.
- I. Sapurina, A. Yu. Osadchev, B. Z. Volchek, M. Trchová, A. Riede and J. Stejskal, Synth. Met., 2002, 129, 29–37 CrossRef CAS.
- J. M. Kinyanjui, R. Harris-Burr, J. G. Wagner, N. R. Wijeratne and D. W. Hatchett, Macromolecules, 2004, 37, 8745 CrossRef CAS.
- I. Xaoier, Cochin University of Science and Technology, Faculty of Technology, Kochi, India, March 2002.
- S. Vyazovkin and N. Sbirrazzuoli, Macromol. Rapid Commun., 2002, 23, 766–770 CrossRef CAS.
- G. Kok, K. Ay, E. Ay, F. Doğan and İ. Kaya, Carbohydr. Polym., 2014, 101, 324–331 CrossRef CAS PubMed.
- F. Doğan, Polym. Eng. Sci., 2014, 54, 992–1002 Search PubMed.
- W. Tang, Y. Liu, X. Yang and C. Wang, Ind. Eng. Chem. Res., 2004, 43, 2054–2059 CrossRef CAS.
- K. Şirin, F. Doğan, M. Balcan and İ. Kaya, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 46, 949–958 Search PubMed.
- F. Doğan, K. Şirin, İ. Kaya and M. Balcan, Polym. Adv. Technol., 2010, 21, 512–519 Search PubMed.
- M. J. Starink, J. Mater. Sci., 2007, 42, 483–489 CrossRef CAS PubMed.
- P. G. Boswell, J. Therm. Anal. Calorim., 1980, 18, 353–358 CrossRef CAS.
- F. Doğan and İ. Kaya, J. Appl. Polym. Sci., 2013, 128, 3782–3793 CrossRef PubMed.
- W. Komatsu, Reactivity of solids, in Proceedings of the 5th International Symposium, Elsevier, Amsterdam, 1965, p. 182 Search PubMed.
- R. Cervini, X. C. Li, G. W. C. Spencer, A. B. Holmes, S. C. Moratti and R. H. Friend, Synth. Met., 1997, 84, 359–360 CrossRef CAS.
- E. T. Seo, R. F. Nelson, J. M. Fritsh, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498–3503 CrossRef CAS.
- F. C. Tsai, C. C. Chang, C. L. Liu, W. C. Chen and S. A. Jenekhe, Macromolecules, 2005, 38, 1958–1966 CrossRef CAS.
- S. Dubey, D. Singh and R. A. Misra, Enzyme Microb. Technol., 1998, 23, 432–437 CrossRef CAS.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. D. Boer, Polym. Rev., 2008, 48, 531–582 CrossRef CAS PubMed.
- X. G. Li, Y. W. Liu, M. R. Huang, S. Peng, L. Z. Gong and M. G. Moloney, Chem.–Eur. J., 2010, 16, 4803–4813 CrossRef CAS PubMed.
- X. G. Li, M. R. Huang, W. Duan and Y. L. Yang, Chem. Rev., 2002, 102, 2925–3030 CrossRef CAS PubMed.
- X. G. Li, M. R. Huang and Y. Yang, Polymer, 2001, 42, 4099–4107 CrossRef CAS.
- W. Luzny and E. Bańka, Macromolecules, 2000, 33, 425–429 CrossRef CAS.
- L. Zhang and M. Wan, Nanotechnology, 2002, 13, 750–755 CrossRef CAS.
- Y. Wang, H. D. Tran, L. Liao, X. Duan and R. B. Kaner, J. Am. Chem. Soc., 2010, 132, 10365–10373 CrossRef CAS PubMed.
- F. R. Diaz, J. Moreno, L. H. Tagle, G. A. East and D. Radic, Synth. Met., 1999, 100, 187–1931 CrossRef CAS.
- İ. Kaya and M. Yıldırım, Eur. Polym. J., 2007, 43, 127–138 CrossRef PubMed.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- L. Qu and G. Shi, Chem. Commun., 2004, 24, 2800–2801 RSC.
- E. Vaganova, M. Rozenberg and S. Yitzchaik, Chem. Mater., 2000, 12, 261–263 CrossRef CAS.
- W. Yang, C. Y. Pan, M. D. Luo and H. B. Zhang, Biomacromolecules, 2010, 11, 1840–1846 CrossRef CAS PubMed.
- F. A. Feist and T. J. Basch, J. Phys. Chem. B, 2008, 112, 9700–9708 CrossRef CAS PubMed.
- B. Lu, J. Xu, C. Fan, F. Jiang and H. Miao, Electrochim. Acta, 2008, 54, 334–340 CrossRef CAS PubMed.
- A. Bilici, F. Dogan, M. Yıldırım and İ. Kaya, J. Phys. Chem. C, 2012, 116, 19934–19940 CAS.
| This journal is © The Royal Society of Chemistry 2015 |