DOI:
10.1039/C5RA07365K
(Paper)
RSC Adv., 2015,
5, 63123-63129
Cobalt aminodiphosphine complexes as catalysts in the oxidation of n-octane†
Received
28th April 2015
, Accepted 8th July 2015
First published on 8th July 2015
Abstract
Two types of cobalt aminodiphosphine complexes have been synthesized and characterized by IR spectroscopy, elemental analyses and single crystal X-ray diffraction. These are [Ph2PN(R)PPh2]CoCl2, 1 and [Ph2P(CH2)2N(R)(CH2)2PPh2]CoCl2, 2 where R = C6H11 (a); C5H11 (b); C3H7 (c). The functional groups on the nitrogen atom (R) were varied from a cyclohexyl ring, to n-pentyl alkyl chain, to an iso-propyl branched substituent. Complexes 2a and 2c were analyzed using single crystal X-ray diffraction. The geometry around the metal centers in 2a and 2c were distorted tetrahedral. All the complexes showed good activity as catalysts for the oxidation of n-octane using tert-butyl hydroperoxide (TBHP) as the oxidant. The complex bearing the flexible ligand backbone with the cyclohexyl substituent on the nitrogen atom was the most active and showed high selectivity towards ketones with 2-octanone being the dominant product.
1. Introduction
The conversion of paraffins or saturated hydrocarbons to more valuable products has drawn the interest of many scientists over the last twenty years.1–3 This is due to the central problem, where general, selective, efficient and catalytic functionalisation reactions of unactivated paraffin C–H bonds remains unsolved.1–10 The need for paraffin activation has practical implications in the replacement of current petrochemical feedstocks (olefins) by economical and easily accessible alkanes, which can result in more efficient strategies for fine chemical synthesis and the proficient use of energy.11–13 However, the conversion of alkanes to desired functionalized products suffers many shortcomings. These include the chemical inertness of the alkanes, the preferential activation of substrates containing sp2 hybridized C–H bonds over sp3 hybridized C–H bonds and cases were the intermediate products are more reactive than the alkane which may react more effortlessly with the metal center.2,3,14–18 Taking into account such shortcomings, a well-suited ligand system is needed. One such system could include the aminodiphosphine ligands. These bi-dentate or multidentate ligands have been used extensively in ethylene oligomerization with chromium as the active metal.19–25 These ligands are ideally suited for catalytic applications in that they are part of a system that displays high activity, stability and variability.26–28 Modification of the ligand backbone, by using different donor substituents or central anionic atoms, tailors the activity of the metals, allowing the reactions of the metal ions to be selective, due to the high demand ligands place on the stereochemistry of the complex.29,30
In this work, a new approach has been undertaken in using cobalt aminodiphosphine complexes in the C–H activation of n-octane. Transition metal-mediated oxidative functionalization of hydrocarbons into useful organic compounds has become an area of immense interest and has led to great advancements in large-scale industrial and synthetic organic processes.31–34 The development of synthetic models has been biologically inspired by a number of enzymes such as methane monooxygenase and cytochrome P450, which make use of a reactive iron–oxo species in the oxidation of a number of alkanes.13,35–39 With the success of first row transition metals, such as iron and copper in the aforementioned enzymes, cobalt serves as promising candidate in the C–H activation of alkanes.40,41 White and co workers have reported site selective C–H activation by a non-heme iron complex in trying to mimic enzymatic activation.3,17,42–44 More recently Tordin and coworkers have used cobalt complexes with tripodal 4N ligands in the oxidation of alkanes, while SNS cobalt complexes have also been studied in the oxidation of n-octane.40,45 Phosphine based ligands have not been widely explored due to ligand degradation or loss of ligand from the metal complex.46 However, Wong and co-workers have used ruthenium based phosphine complexes in the oxidation of n-octane and reported low conversions.47,48 These catalytic oxidation processes are carried out using a variety of oxidants, namely, PhIO, NaOCl, H2O2, alkyl hydroperoxides, percarboxylic acids and molecular oxygen.33,49–52
We herein report the synthesis and characterization of two sets of cobalt aminodiphosphine complexes (1 and 2) and their application in the catalytic oxidation of n-octane in acetonitrile using tert-butyl hydroperoxide (TBHP) as the oxidant (Fig. 1). The rigidity (1) and flexibility (2) of the ligand backbone was varied to asses whether this will influence the catalytic activity. The substituent on the nitrogen atom was also varied making use of three different types of functional groups, a ringed (cyclohexyl), straight chained (n-pentyl) and a branched (iso-propyl) substituent with the intention of observing if these groups have an effect on the catalytic activity and selectivity to the products of oxidation. To our knowledge the complexes used in this study are new. Furthermore, limited studies have been carried out in the activation of n-alkanes, as compared to cycloalkanes, due to their low activity and tendency to undergo over oxidation. However, valorization of medium length chain n-alkanes is of special importance since these are the building blocks in the chemical industry and provide a cheaper alternate feedstock.53–56
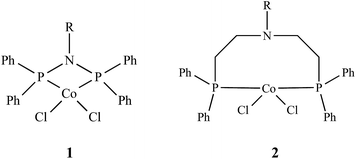 |
| | Fig. 1 Representation of complexes 1 and 2; R = cyclohexyl for 1a or 2a; n-pentyl for 1b or 2b and iso-propyl for 1c or 2c. | |
2. Experimental
2.1 Synthesis and characterization of the compounds
All experiments were performed using standard Schlenk techniques under inert conditions in moisture free glassware with anhydrous solvents. All solvents were analytical grade. To render the reaction glassware moisture free, it was heated with a heat gun followed by cycles of vacuum and nitrogen pressure. Diethyl ether and hexane were distilled from sodium benzophenoneketyl under nitrogen. Dichloromethane was distilled from P2O5, and ethanol from magnesium turnings. Deuterated solvents were used as received and stored in a desiccator. The NMR spectra were recorded at 400 MHz (1H), 100 MHz (13C) and 162 MHz (31P) using a Bruker Ultrashield 400 MHz spectrometer. 1H NMR and 13C{1H} NMR chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane. 1H NMR and 13C{1H} NMR signals were referenced to the residual hydrogen signal of CDCl3 (7.26 ppm) and (77.16 ppm) respectively. 31P NMR chemical shifts were reported in parts per million (ppm) from triphenylphosphine (−17.6 ppm). The FT-IR spectra were recorded using a Perkin Elmer Universal Attenuated Total Reflection (ATR) Sampling Accessory attached to the FT-IR series 100. Elemental analyses were carried out on a Thermo-Scientific Flash 2000 CHNS/O analyzer. All PNP (1 and 2) ligands were synthesized with modification of literature procedure.20,57
Synthesis of [Ph2PN(Cy)PPh2]CoCl2 (1a). The synthesis was adapted from a reported procedure in literature.58 To a 100 ml two necked round bottom flask, 10 ml of ethanol was added and purged with nitrogen for 10 minutes. Thereafter, [Ph2PN(Cy)PPh2] (0.62 mmol, 0.29 g) and CoCl2·6H2O (0.63 mmol, 0.15 g) were added. The solution was slowly stirred at room temperature. The round bottom flask was equipped with a condenser and the solution was brought to reflux at 77 °C. After 15 minutes under reflux the solution changed color from blue to green. After 24 hours diethyl ether was added to allow precipitation of the complex. The solvent was decanted and the complex was washed with diethyl ether (3 × 10 ml) and dried under high vacuum. Crystals were grown from diethyl ether and dichloromethane through vapour diffusion. Yield: 59%, 0.22 g. Decomposes > 192 °C. IR νmax (ATR)/cm−1: 997 (m), (P–N); 1067 (m) (cyclohexyl ring vibrations); 1433 (m) (aromatic ring); 2852 (s) (CH2). Anal. (%) calcd for C30H31Cl2CoNP2: C: 60.3%; H: 5.2%; N: 2.3%. Found: C: 60.3%; H: 5.2%; N: 2.3%.
Synthesis of [Ph2PN(C5H11)PPh2]CoCl2 (1b). Synthesized according to the procedure described for 1a except that [Ph2PN(C5H11)PPh2] (0.62 mmol, 0.290 g) was used. Yield: 45%, 0.16 g. Decomposes > 227 °C. IR νmax (ATR)/cm−1: 998 (s), (P–N); 1434 (m) (aromatic ring); 2945 (s) (CH2). Anal. (%) calcd for C29H31Cl2CoNP2: C: 59.5%; H: 5.3%; N: 2.4%. Found: C: 58.6%; H: 6.0%; N: 2.2%.
Synthesis of [Ph2PN(C3H7)PPh2]CoCl2 (1c). Synthesized according to the procedure described for 1a except that [Ph2PN(C3H7)PPh2] (0.62 mmol, 0.27 g) was used. Yield: 61%, 0.21 g. Decomposes > 165 °C. IR νmax (ATR)/cm−1: 998 (s), (P–N); 1434 (m) (aromatic ring); 2933 (s) (CH2). Anal. (%) calcd for C27H27Cl2CoNP2: C: 58.2%; H: 4.9%; N: 2.5%. Found: C: 58.0%; H: 5.2%; N: 2.4%.
Synthesis of [Ph2PC2H4N(Cy)C2H4PPh2]CoCl2 (2a). Synthesized according to the procedure described for 1a except that [Ph2PC2H4N(Cy)C2H4PPh2] (0.62 mmol, 0.32 g) was used. Yield: 81%, 0.33 g. Melting point: 251–252 °C. IR νmax (ATR)/cm−1: 1030 (m) (cyclohexyl ring vibrations); 1434 (m) (aromatic ring); 2931 (s) (CH2). Anal. (%) calcd for C34H39Cl2CoNP2: C: 62.5%; H: 6.02%; N: 2.14%. Found: C: 61.8%; H: 6.08%; N: 2.08%.
Synthesis of [Ph2PC2H4N(C5H11)C2H4PPh2]CoCl2 (2b). Synthesized according to the procedure described for 1a except that [Ph2PC2H4N(C5H11)C2H4PPh2] (0.62 mmol, 0.32 g) was used. Yield: 82%, 0.26 g. Melting point: 190–192 °C. IR νmax (ATR)/cm−1: 1434 (m) (aromatic ring); 2952 (s) (CH2). Anal. (%) calcd for C34H39Cl2CoNP2: C: 61.8%; H: 6.13%; N: 2.18%. Found: C: 62.1%; H: 6.18%; N: 2.18%.
Synthesis of [Ph2PC2H4N(C3H7)C2H4PPh2]CoCl2 (2c). Synthesized according to the procedure described for 1a except that [Ph2PC2H4N(C3H7)C2H4PPh2] (0.62 mmol, 0.31 g) was used. Yield: 40%, 0.15 g. Melting point: 250–253 °C. IR νmax (ATR)/cm−1: 1435 (m) (aromatic ring); 2869 (s) (CH2); 2964 (s) (CH). Anal. (%) calcd for C31H35Cl2CoNP2: C: 60.7%; H: 5.75%; N: 2.28%. Found: C: 60.4%; H: 5.83%; N: 2.19%.
2.2 Crystal structure analysis
Crystals of compounds 2a and 2c were grown by the vapour diffusion of diethyl ether into a solution of the complexes in dichloromethane at room temperature to give blue crystals for 2a and 2c. The crystals of the complexes were each selected and glued onto the tip of glass fibers separately. The crystals were then mounted in a stream of cold nitrogen at 100(1) K and centered in the X-ray beam by using a video camera. Crystal evaluation and data collection were performed on a Bruker Smart APEXII diffractometer with Mo Kα radiation (λ = 0.71073 Å) and a diffractometer to crystal distance of 4.00 cm. The initial cell matrix was obtained from three series of scans at different starting angles. Each series consisted of 12 frames collected at intervals of 0.5° in a 6° range with the exposure time of about 10 seconds per frame. The reflections were successfully indexed by an automated indexing routine built in the APEXII program suite.59 Data collection method involved ω scans of width 0.5°. Data reduction was carried out using the program SAINT+.59 The structures were solved by direct methods using SHELXS60 and refined by SHELXL.59 All structures were checked for solvent-accessible cavities using PLATON61 and the graphics were performed with ORTEP3.
Non-H atoms were first refined isotropically and then by anisotropic refinement with full-matrix least-squares calculations based on F2 using SHELXS.61 All H atoms were positioned geometrically and allowed to ride on their respective parent atoms. All H atoms were refined isotropically. Crystal data and structure refinement information for all the complexes are summarized in Table 1.†
Table 1 Crystal data and structure refinement for complexes 2a and 2c
| |
2a |
2c |
| Empirical formula |
C34H39Cl2CoNP2 |
C31H35Cl2CoNP2 |
| Formula weight |
653.43 |
613.37 |
| Temperature K |
173(2) |
173(2) |
| Wavelength Å |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
| a (Å) |
9.7475(16) |
9.1436(3) |
| b (Å) |
11.1289(18) |
16.8343(5) |
| c (Å) |
30.195(5) |
19.0478(6) |
| α (°) |
90 |
90 |
| β (°) |
104.044(9) |
95.1650(10) |
| γ (°) |
90 |
90 |
| Volume (Å3) |
3177.6(9) |
2920.05(16) |
| Z |
4 |
4 |
| Densitycalc Mg/m3 |
1.366 |
1.395 |
| Absorption coefficient mm−1 |
0.833 |
0.902 |
| F(000) |
1364 |
1276 |
| Crystal size (mm3) |
0.19 × 0.11 × 0.11 |
0.19 × 0.11 × 0.11 |
| Theta range (°) |
1.96 to 25.00 |
2.15 to 25.00 |
| Index ranges |
−11 ≤ h ≤ 11; −13 ≤ k ≤ 13; −30 ≤ l ≤ 35 |
−10 ≤ h ≤ 10; −20 ≤ k ≤ 20; −22 ≤ l ≤ 22 |
| Independent reflections |
5547 [R(int) = 0.1299] |
5132 [R(int) = 0.0314] |
| Completeness to theta |
25.00°; 99.1% |
100.0 |
| Max. and min. trans |
0.9139 and 0.8577 |
0.9073 and 0.8473 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
5547/0/361 |
5132/0/336 |
| Goodness-of-fit on F2 |
1.170 |
1.025 |
| Final R indices [I > 2σ(I)] |
R1 = 0.1014, wR2 = 0.2331 |
R1 = 0.0229, wR2 = 0.0496 |
| R indices (all data) |
R1 = 0.1375, wR2 = 0.2449 |
R1 = 0.0301, wR2 = 0.0534 |
| Largest diff. peak and hole (e Å−3) |
1.035 and −0.804 |
0.364 and −0.268 |
2.3 Oxidation of n-octane
All catalytic reactions were performed under inert conditions in moisture free glassware with anhydrous solvents. MeCN was degassed for 10–15 minutes before use. All reagents were weighed and handled in air. All products were analyzed using a PerkinElmer Auto System gas chromatograph fitted with a Flame Ionisation Detector (FID) set at 260 °C. A Pona column (50 m × 0.20 mm × 0.5 μm) was utilized with the injector temperature set at 240 °C. Catalytic testing was carried out in acetonitrile at 80 °C, using tert-butyl hydroperoxide (TBHP) as the respective oxidant. The catalyst to substrate ratio was kept constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. A two-necked pear shaped flask was charged with 10 mg of the respective catalyst, pentanoic acid (as an internal standard), n-octane, TBHP and 10 ml of the solvent. The flask was equipped with a reflux condenser. The mixture was stirred, heated to the respective temperature and maintained for 48 hours in an oil bath. After the time period, an aliquot was removed using a Pasteur pipette and filtered through cotton wool and a silica gel plug, after which PPh3 was added (for reduction of the remaining TBHP and alkylperoxides which are formed as primary products in alkane oxidation).33 An aliquot (0.5 μl) was injected into the GC and quantified.
:100. A two-necked pear shaped flask was charged with 10 mg of the respective catalyst, pentanoic acid (as an internal standard), n-octane, TBHP and 10 ml of the solvent. The flask was equipped with a reflux condenser. The mixture was stirred, heated to the respective temperature and maintained for 48 hours in an oil bath. After the time period, an aliquot was removed using a Pasteur pipette and filtered through cotton wool and a silica gel plug, after which PPh3 was added (for reduction of the remaining TBHP and alkylperoxides which are formed as primary products in alkane oxidation).33 An aliquot (0.5 μl) was injected into the GC and quantified.
3. Results and discussion
3.1 Synthesis and characterization of the compounds
Complexes 1 and 2 were synthesized by adaptation of a procedure by Romerosa et al.58 The respective ligands were added to a solution of CoCl2·6H2O in ethanol and after refluxing for 24 hours, diethyl ether was added to allow precipitation of the respective complex. The precipitates of complexes 1 were green, whilst those of 2 were blue. The complexes where fully characterized by elemental analyses, infrared spectroscopy and single crystal X-ray diffraction. The complexes are paramagnetic, hence elucidation by NMR was unsuccessful.58 The elemental analyses of the complexes matched the calculated values and this is indicative of complexation, as are the sharp melting points and crystal structures of 2a and 2c. A shift in the νP–N band in the IR spectra of complexes 1 and their respective ligands also are noted and shown in Table 2.
Table 2 Comparison of the νP–N band shifts of complexes 1 and their respective ligands
| Substituent |
νP–N (Ligand)/cm−1 |
νP–N (Complex 1)/cm−1 |
| a |
982 |
997 |
| b |
977 |
998 |
| c |
986 |
998 |
3.2 Description of crystal structures
Blue crystals of complexes 2a and 2c were obtained by vapour diffusion of diethyl ether into a dichloromethane solutions of complexes 2a and 2c. ORTEP diagrams of 2a and 2c are given in Fig. 2, while bond distances and bond angles are provided in Table 3. Both compounds crystallize with one molecule of the respective complexes in the asymmetric units. In both complexes the cobalt metal is bound to one ligand through the phosphorous atoms of the ligand and two chlorine atoms, resulting in a distorted tetrahedral geometry around the metal center. The P–Co–P bond angles are 116.42(9) and 116.32(2)° in 2a and 2c respectively, while the two Cl–Co–P angles lie between 100.53(7) and 108.35(1)°. The Cl–Co–Cl bond angles are 120.4(1) and 118.50(2)° in 2a and 2c respectively. The P–Co bond lengths for both complexes are comparable to related complexes found in literature.62
 |
| | Fig. 2 The molecular structures of complexes 2a and 2c showing part of the atom-numbering scheme. Displacement ellipsoids are drawn at 50% probability level and H atoms have been omitted for clarity. | |
Table 3 Selected bond lengths (Å) and angles (°) for complexes 2a and 2c
| 2a |
2c |
| Bond lengths |
| Co(1)–P(1) |
2.348(3) |
Co(1)–P(1) |
2.3618(5) |
| Co(1)–P(2) |
2.368(3) |
Co(1)–P(2) |
2.3684(5) |
| Co(1)–Cl(1) |
2.225(3) |
Co(1)–Cl(1) |
2.2363(5) |
| Co(1)–Cl(2) |
2.245(3) |
Co(1)–Cl(2) |
2.2169(5) |
|
| Bond angles |
| P(1)–Co(1)–P(2) |
116.42(9) |
P(1)–Co(1)–P(2) |
116.32(2) |
| Cl(1)–Co(1)–Cl(1) |
120.39(10) |
Cl(1)–Co(1)–Cl(1) |
118.50(2) |
| Cl(1)–Co(1)–P(1) |
106.15(7) |
Cl(2)–Co(1)–P(2) |
103.357(18) |
| Cl(2)–Co(1)–P(2) |
102.53(9) |
Cl(1)–Co(1)–P(1) |
108.346(18) |
| Cl(2)–Co(1)–P(1) |
100.53(7) |
Cl(2)–Co(1)–P(1) |
102.530(18) |
| Cl(1)–Co(1)–P(2) |
110.99(10) |
Cl(1)–Co(1)–P(2) |
108.116(18) |
3.3 Oxidation of n-octane
The catalytic activity of complexes 1 and 2 were explored in the oxidation of n-octane. All catalytic runs were carried out at 80 °C in acetonitrile with TBHP (t-BuOOH) as the oxidant under argon atmosphere. Preliminary work showed low conversions with no significant change in product selectivity at lower temperatures. TBHP as an oxidant has been used in a number of oxidation reactions and has the advantage over other oxidants in that it has higher solubility in organic solvents, which contain dissolved hydrophobic hydrocarbons.63 Optimization of the substrate to oxidant ratio was carried out, by investigating n-octane to TBHP ratios of 1:2.5; 1:5; 1:7.5 and 1:10, where the ratio of 1:5 gave the highest conversion with good selectivity. Control experiments were carried out in the absence of the catalyst and oxidant respectively. In the former, a 2% conversion was observed with the highest selectivity to 2-octanone (Fig. 3). However, the latter reaction showed a 0% conversion. Testing was also carried out with cobalt chloride (CoCl2·6H2O) under the same catalytic conditions. A 2% conversion was observed, with selectivity to the over oxidized products, namely the ketones and octanoic acid. This is the same conversion as for the blank reaction with TBHP only, but with a greater selectivity to the over oxidized products.
 |
| | Fig. 3 Selectivity of the blank reaction (no catalyst) to products of oxidation at a ratio of 1:5 of substrate to oxidant at 80 °C in acetonitrile. | |
The more sterically hindered, more rigid, catalysts 1 show more limited activity in comparison to the more flexible catalysts 2, which have a significantly larger bite angle. The bite angle is known to have an impact on the activity and selectivity of catalytic reactions.64 It has been reported that the effect of bite angle on C–X bond activation originates from an electronic factor, where the donor–acceptor orbital interactions (metal d orbitals to the substrate σ*C–X) stabilize the transition state.65 As the metal–ligand d-hybrid orbital is driven to smaller bite angles, the transition state becomes more stabilized.65 Interestingly, a decrease in the activity for both catalysts 1 and 2 is observed, as one moves from the cyclohexyl (a) to the iso-propyl (c) substituent (Fig. 4). Noteworthy, for these ligands used in ethylene tetramerisation, a similar trend is observed in terms of their activity with chromium as the active metal.20 This can be attributed to the basicity of the substituent on the nitrogen atom.20
 |
| | Fig. 4 Conversion of n-octane by catalysts 1 and 2 at a ratio of 1:5 of substrate to oxidant at 80 °C in acetonitrile. | |
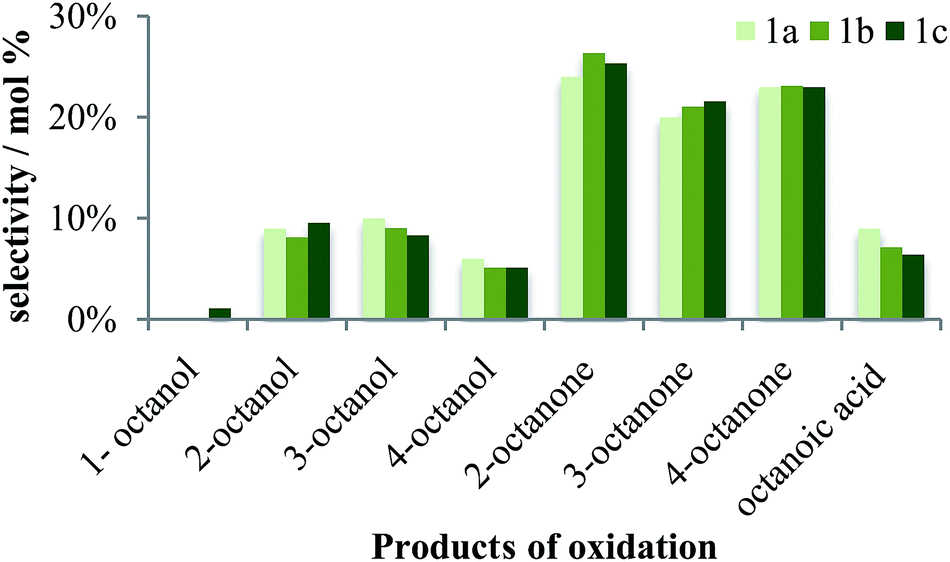
Both sets of catalysts (1 and 2) are highly selective to the ketones, with 2-octanone being the dominant product (Fig. 5 and 6). This indicates that the catalyst causes oxidation of the internal carbons more readily than the terminal carbon. Such cases, with 2-octanone being the dominant product, have been observed in the biological hydroxylation of alkanes catalyzed by methane monooxygenase.13 The C(2) position is three times as active as the C(1), and is most reactive in linear alkane chains as reflected by the regioselectivity in reactions for n-heptane, as well as n-octane.5,51,63,66,67 Thus, early work by Saussine and co-workers showed that cobalt(III)alkylperoxy complexes also were highly selective to the 2-octanone in the oxidation of n-octane.68 It has been shown with sandwich type tungstophosphate anions, [M4(H2O)2(PW9O34)2]10−, where M = Co2+, Mn2+ and Fe2+, that highest selectivity to the ketone product is observed when cobalt salts were used, in comparison to using the manganese and iron salts, in the oxidation of cyclohexane.69 Ketone formation is also observed with ruthenium phosphine compounds, in which cases only 2- and 4-octanone are observed with a 4% conversion.47,48 In addition, in the oxidation of lower chain alkanes, such as heptane and propane, using ruthenium complexes as catalysts, higher yields to the ketonic products and very low yields to the alcohols were observed.70,71 Chen and White have reported yields to 2- and 3-octanone with no selectivity to primary products using a bulky iron electrophilic catalyst.3 Over-oxidation is highly prevalent at the C(1) position of the n-octane chain for both catalysts 1 and 2, with higher selectivity to octanoic acid and very low selectivity to 1-octanol and no selectivity to octanal. Assuming a sequential oxidation, this implies that octanal is highly reactive over these catalysts.
 |
| | Fig. 5 Selectivity of catalyst 1 to the products of oxidation at a ratio of 1:5 of substrate to oxidant at 80 °C in acetonitrile. | |
 |
| | Fig. 6 Selectivity of catalyst 2 the products of oxidation at a ratio of 1:5 of substrate to oxidant at 80 °C in acetonitrile. | |
Following the method of Shul'pin and coworkers, addition of triphenylphosphine to a filtered (through a plug of silica to remove the catalyst) aliquot of the reaction mixture 10 min prior to GC analysis was performed, since a relative increase in the alcohol peak and a decrease in the ketone peak can result.33 This was the true concentration of the alcohols and ketones, since the alkyl hydroperoxides that are present are completely reduced to the corresponding alcohols. For catalysts 1, a change in the selectivity to the alcohol and ketone before and after addition of the PPh3 was observed, however, for catalysts 2 no observable change was noted.
The regioselectivity parameters (Table 4) further indicate that the C(2) position is the most activated carbon of the n-octane chain, with the C(1) being the least activated. The catalysts with the pentyl and iso-propyl (b and c) substituents are most selective to the alcohols, with catalysts 2 being more selective to the alcohols than 1.
Table 4 Selectivity parameters in the oxidation of n-octane by catalysts 1 and 2
| Catalyst |
Alcohola C(1):C(2):C(3):C(4) |
Ketoneb C(2):C(3):C(4) |
Totalc C(1):C(2):C(3):C(4) |
| Parameters C(1):C(2):C(3):C(4) are the relative reactivities of hydrogen atoms at carbon 1, 2, 3 and 4 of the n-octane chain. The calculated reactivities from the % selectivity are normalized, i.e. calculated taking into account the number of hydrogen atoms at each carbon. Includes the % selectivity of octanoic acid, alcohols and ketones and the values are normalized. |
| 1a |
0:1.5:1.7:1 |
1.2:1:1.2 |
1:5.5:5:4.8 |
| 1b |
0:1.6:1.8:1 |
1.2:1:1.1 |
1:7.4:6.5:6.1 |
| 1c |
0:2:1.6:1 |
1.1:1:1.0 |
1:7.6:6.5:6.1 |
| 2a |
1:0:3.3:3.3 |
1.4:1:1 |
1:5.2:4.1:4.2 |
| 2b |
1:4.2:4.2:2.9 |
1.1:1:1 |
1:4:3.5:3.4 |
| 2c |
1:4.2:2.9:2.9 |
1.2:1:1 |
1:4.1:3.4:3.5 |
When using TBHP as an oxidant, the ketone product forms from the oxidation of the alcohol (over oxidation).35 Since the selectivity to the ketones is much greater, it is likely that the oxidation reaction proceeds via the formation of hydroxyl radicals where the metal complex activates the oxidant, t-BuOOH, forming a reactive species, the hydroxyl radical, which attacks n-octane.5,31,72–75 We assume that the reaction takes place in the coordination sphere of the metal complex which would explain the influence of the ligand system on the reaction. t-BuO˙ is proposed to form by the reduction of t-BuOOH by the Co(II)L which generates a hydroxo–Co(III) species.73,74 This specie reacts further with t-BuOOH regenerating Co(II)L,73,74 with t-BuO˙ and O2. The t-BuO˙ attacks the n-octane forming octyl radicals, R˙, which react with oxygen to form octyl peroxy radicals, ROO˙.73,74 These react with the n-octane to form the octyl-hydroperoxide ROOH, which undergoes homolytic decomposition to form the organooxyl radical, RO˙. By H-abstraction from n-octane, the RO˙ radical forms octanols (ROH), which can react further with oxygen to form the octanones.73,74 Isolation and characterization of the used catalysts was unsuccessful despite many attempts.
4. Conclusions
In this paper, new cobalt aminodiphosphine complexes have been synthesized and fully characterized and were used as catalysts in the oxidation of n-octane. Higher conversion is seen with the more flexible complexes 2 as compared to the more sterically hindered complexes 1 and this may be attributed to their bite angle. The substituent on the nitrogen atom has an effect of the activity, where the catalysts with the cyclohexyl ring substituents are much more active than those with branched or straight chain substituents. The ketones were the dominant product, with the C(2) position being the most activated in the octane chain contributing to the high selectivity of 2-octanone (34%). 1-Octanol was prominent with catalysts 2, however, over oxidation was also more evident with these catalysts contributing to the high selectivity of the ketones.
Acknowledgements
The authors gratefully acknowledge the financial support from NRF, THRIP (Grant number 1208035643) and UKZN (URF).
References
- R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083 CrossRef CAS PubMed.
- M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2010, 327, 566 CrossRef CAS PubMed.
- Y. He, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2011, 50, 12651 CrossRef CAS PubMed.
- A. M. Kirillov, M. V. Kirillova, L. S. Shul'pina, P. J. Figiel, K. R. Gruenwald, M. F. C. Guedes da Silva, M. Haukka, A. J. L. Pombeiro and G. B. Shul'pin, J. Mol. Catal. A: Chem., 2011, 350, 26 CrossRef CAS PubMed.
- M. V. Kirillova, A. M. Kirillov, D. Mandelli, W. A. Carvalho, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2010, 272, 9 CrossRef CAS PubMed.
- B. L. Conley, W. J. Tenn III, K. J. H. Young, S. K. Ganesh, S. K. Meier, V. R. Ziatdinov, O. Mironov, J. Oxgaard, J. Gonzales, W. A. Goddard III and R. A. Periana, J. Mol. Catal. A: Chem., 2006, 251, 8 CrossRef CAS PubMed.
- M. N. Kopylovich, A. M. Kirillov, A. K. Baev and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2003, 206, 163 CrossRef CAS.
- A. Sivaramakrishna, P. Suman, G. E. Veerashekhar, S. Janardan, C. Sravani, T. Sandeep, K. Vijayakrishna and H. S. Clayton, J. Coord. Chem., 2013, 66, 2091 CrossRef CAS PubMed.
- R. D. Young, Chem.–Eur. J., 2014, 20, 12704 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed.
- S. R. Klei, K. L. Tan, J. T. Golden, C. M. Yung, R. K. Thalji, K. A. Ahrendt, J. A. Ellman, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2004, 885, 46 CAS.
- A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Coord. Chem. Rev., 2012, 256, 2741 CrossRef CAS PubMed.
- A. M. Kirillov and G. B. Shul'pin, Coord. Chem. Rev., 2013, 257, 732 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS PubMed.
- K. J. Fraunhoffer, D. A. Bachovchin and M. C. White, Org. Lett., 2004, 7, 223 CrossRef PubMed.
- N. A. Vermeulen, M. S. Chen and M. C. White, Tetrahedron, 2009, 65, 3078 CrossRef CAS PubMed.
- B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391 CrossRef CAS PubMed.
- S. Teo, Z. Weng and T. S. A. Hor, Organometallics, 2008, 27, 4188 CrossRef CAS.
- K. Blann, A. Bollmann, H. de Bod, J. T. Dixon, E. Killian, P. Nongodlwana, M. C. Maumela, H. Maumela, A. E. McConnel, D. H. Morgan, M. J. Overette, M. Pŕetorius, S. Kuhlmann and P. Wasserscheid, J. Catal., 2007, 249, 244 CrossRef CAS PubMed.
- K. Blann, A. Bollmann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. H. Morgan, A. Neveling, S. Otto and M. J. Overett, Chem. Commun., 2005, 620 RSC.
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuiness, D. H. Morgan, A. Neveling, S. Otton, M. Overette, A. M. Z. Slawin, P. Wassercheid and S. Kihlmann, J. Am. Chem. Soc., 2004, 126, 14712 CrossRef CAS PubMed.
- M. J. Overett, K. Blann, A. Bollmann, J. T. Dixon, F. Hess, E. Killian, H. Maumela, D. H. Morgan, A. Neveling and S. Otto, Chem. Commun., 2005, 622 RSC.
- L. E. Bowen, M. F. Haddow, A. G. Orpen and D. F. Wass, Dalton Trans., 2007, 1160 RSC.
- L. E. Bowen, M. Charernsuk, T. W. Hey, C. L. McMullin, A. G. Orpen and D. F. Wass, Dalton Trans., 2010, 560 RSC.
- D. Benito-Garagorri, L. G. a. Alves, M. Puchberger, K. Mereiter, L. F. Veiros, M. J. Calhorda, M. D. Carvalho, L. P. Ferreira, M. Godinho and K. Kirchner, Organometallics, 2009, 28, 6902 CrossRef CAS.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201 CrossRef CAS PubMed.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed.
- X. Xu, Z. Xi, W. Chen and D. Wang, J. Coord. Chem., 2007, 60, 2297 CrossRef CAS PubMed.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 CrossRef CAS.
- G. B. Shul'pin, C. R. Chim., 2003, 6, 163 CrossRef.
- K. C. Gupta, A. Kumar Sutar and C. C. Lin, Coord. Chem. Rev., 2009, 253, 1926 CrossRef CAS PubMed.
- G. B. Shul'pin, A. R. Kudinov, L. S. Shul'pina and E. A. Petrovskaya, J. Organomet. Chem., 2006, 691, 837 CrossRef PubMed.
- S. S. Stahl, J. A. Labinger and J. E. Bercaw, Angew. Chem., Int. Ed., 1998, 37, 2180 CrossRef.
- S. Yiu, W. Man and T. Lau, J. Am. Chem. Soc., 2008, 130, 10821 CrossRef CAS PubMed.
- G. V. Nizova, B. Krebs, G. Süss-Fink, S. Schindler, L. Westerheide, G. L. Cuervo and G. B. Shul'pin, Tetrahedron, 2002, 58, 9231 CrossRef CAS.
- B. Bahramian, V. Mirkhani, M. Mogahadam and S. Tangestaninejad, Catal. Commun., 2006, 7, 289 CrossRef CAS PubMed.
- F. G. Doro, J. R. L. Smith, A. G. Ferreira and M. D. Assis, J. Mol. Catal. A: Chem., 2000, 164, 97 CrossRef CAS.
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738 CrossRef CAS PubMed.
- E. Tordin, M. List, U. Monkowius, S. Schindler and G. Knör, Inorg. Chim. Acta, 2013, 402, 90 CrossRef CAS PubMed.
- W. Kanjina and W. Trakarnpruk, J. Met., Mater. Miner., 2010, 20, 29 CAS.
- P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052 CrossRef CAS PubMed.
- M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721 CrossRef CAS PubMed.
- M. C. White, Synlett, 2012, 2746 CAS.
- L. Soobramoney, M. D. Bala and H. B. Friedrich, Dalton Trans., 2014, 15968 RSC.
- G. J. P. Britovsek, J. England, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Dalton Trans., 2005, 945–955 RSC.
- W. K. Wong, X. P. Chen, T. W. Chik, W. Y. Wong, J. P. Guo and F. W. Lee, Eur. J. Inorg. Chem., 2003, 3539 CrossRef CAS PubMed.
- W. K. Wong, X. P. Chen, J. P. Guo, Y. G. Chi, W. X. Pan and W. Y. Wong, J. Chem. Soc., Dalton Trans., 2002, 1139 RSC.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and N. Rasouli, Catal. Commun., 2008, 9, 2411 CrossRef CAS PubMed.
- A. Cagnina, S. Campestrini, F. Di Furia and P. Ghiotti, J. Mol. Catal. A: Chem., 1998, 130, 221 CrossRef CAS.
- L. J. R. Smith, Y. Iamamoto and F. S. Vinhado, J. Mol. Catal. A: Chem., 2006, 252, 23 CrossRef PubMed.
- R. Pereira, M. Rufo and U. Schuhardt, J. Braz. Chem. Soc., 1994, 5, 83 CrossRef CAS.
- N. Gounden, H. B. Friedrich, N. Mahadevaiah and M. I. Fadlalla, Catal. Lett., 2014, 144, 2043 CrossRef CAS.
- V. D. B. C. Dasireddy, S. Singh and H. B. Friedrich, J. Mol. Catal. A: Chem., 2014, 395, 398 CrossRef CAS PubMed.
- V. D. B. C. Dasireddy, H. B. Friedrich and S. Singh, Appl. Catal., A, 2013, 467, 142 CrossRef CAS PubMed.
- M. Naraynappa, V. D. B. C. Dasireddy and H. B. Friedrich, Appl. Catal., A, 2012, 447–448, 135 CrossRef PubMed.
- K. K. Hii, M. Thornton-Pett, A. Jutand and R. P. Tooze, Organometallics, 1999, 18, 1887 CrossRef CAS.
- A. Romerosa, C. Saraiba-Bello, M. Serrano-Ruiz, A. Caneschi, V. McKee, M. Peruzzini, L. Sorace and F. Zanobini, Dalton Trans., 2003, 3233 RSC.
- Bruker, SAINT-plus, APEXII, SHELXL, SADABS, Bruker-AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- Q. Dong, M. J. Rose, W.-Y. Wong and H. B. Gray, Inorg. Chem., 2011, 50, 10213 CrossRef CAS PubMed.
- G. B. Shul'pin, Mini-Rev. Org. Chem., 2009, 6, 95 CrossRef.
- P. W. N. M. van Leeuwen, P. C. J. Kamer and J. N. H. Reek, Pure Appl. Chem., 1999, 71, 1443 CrossRef CAS.
- W.-J. van Zeist, R. Visser and F. M. Bickelhaupt, Chem.–Eur. J., 2009, 15, 6112 CrossRef CAS PubMed.
- A. J. Bailey, W. P. Griffith and P. D. Savage, J. Chem. Soc., Dalton Trans., 1995, 3537 RSC.
- G. B. Shul'pin, G. Süss-Fink and L. S. Shul'pina, J. Mol. Catal. A: Chem., 2001, 170, 17 CrossRef.
- L. Saussine, E. Brazi, A. Robine, H. Mimoun, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1985, 107, 3534 CrossRef CAS.
- I. C. M. S. Santos, J. A. F. Gamelas, M. S. S. Balula, M. M. Q. Simões, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2007, 262, 41 CrossRef CAS PubMed.
- T. C. Lau and C. K. Mak, J. Chem. Soc.,
Chem. Commun., 1995, 9, 943 RSC.
- S. Murahashi, T. Saito, H. Hanaoka, Y. Murakami, T. Naota, H. Kumobayashi and S. Akutagawa, J. Org. Chem., 1993, 58, 2929 CrossRef CAS.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS PubMed.
- T. C. O. Mac Leod, M. V. Kirillova, A. J. L. Pombeiro, M. A. Schiavon and M. D. Assis, Appl. Catal., A, 2010, 372, 191 CrossRef CAS PubMed.
- S. Förster, A. Rieker, K. Maruyama, K. Murata and A. Nishinaga, J. Org. Chem., 1996, 61, 3320 CrossRef.
- G. B. Shul'pin, C. C. Golfeto, G. Süss-Fink, L. S. Shul'pina and D. Mandelli, Tetrahedron Lett., 2005, 46, 4563 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.