
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrophilic trifluoromethylthiolation of thiols with trifluoromethanesulfenamide†
Marjan
Jereb
* and
Darko
Dolenc
Department of Organic Chemistry, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1001 Ljubljana, Slovenia. E-mail: marjan.jereb@fkkt.uni-lj.si; Fax: +386 1 241 9144; Tel: +386 1 479 8577
First published on 26th June 2015
Abstract
The highly selective and effective, metal-free, acid promoted trifluoromethylthiolation of thiols to the corresponding trifluoromethyl disulfides is described. The aryl-, benzyl-, aliphatic-, and heteroaromatic thiols reacted selectively, thus proving excellent reaction generality. The method offers practical and easy access to the previously mostly unknown or rarely reported trifluoromethyl disulfides. Comparison of the relative reactivity of thiophenols suggests formation of an electron-deficient intermediate in the transition state, which was supported by quantum chemical calculations. The supposed reaction course is discussed.
Introduction
Fluorinated organic molecules have been gaining significance in different fields i.e. medicinal and agrochemistry, high-performance advanced materials and new reaction media such as fluorinated ionic liquids and perfluorinated solvents.1 The fluorine atom in an organic molecule brings about several stereoelectronic changes, and the C–F bond is an important conformational and bioisosteric tool in bioorganic chemistry.2 The fluorine atom is important in inter- and intramolecular interactions; moreover, it is of particular significance in molecular recognition3 and crystal engineering.4 The trifluoromethyl group is one of the strategic fluorine-containing substituents and there has been immense interest in its introduction into organic molecules.5,6 The discovery of the first electrophilic CF3-transfer agent, by Yagupolskii in 1984,7 induced new developments in this field.8,9 The introduction of the trifluoromethyl group into organic molecules using nucleophilic,10 radical11 and metal-based12 CF3 sources has also progressed very rapidly.A very attractive modulation of the CF3– group is CF3S– trifluoromethylthiol group, although its introduction have been studied much less than that of the CF3– group. The trifluoromethylthiol group CF3S– exhibits a remarkably high lipophilicity parameter, and is one of the key fragments in certain biologically relevant compounds.13 The 2′-SCF3 substituted uridine derivative was found to be a potent label for probing structure and function of RNA by 19F NMR spectroscopy.14 There are different ways of introduction of CF3S– functionality. The introduction of the CF3S– group could be performed directly,15 by interconversion of functional groups16 or by trifluoromethylation of suitable sulfur-containing moieties.17 Direct methods usually rely on heavy-metal-based reagents18 and/or catalysts or on the use of extremely toxic and hazardous trifluoromethyldisulfide19 or trifluoromethylsulfenyl chloride.20 Recently, considerable steps forward have been made in the direct introduction of the CF3S– functionality.21 Several new SCF3 transfer agents have been developed, particular of an electrophilic nature. The recent progress was initiated by the work of Billard, Langlois and coworkers when they published a synthesis of PhNHSCF31 and its derivatives.22 These compounds are easy-to-handle electrophilic SCF3 reagents that can react with alkenes and alkynes,23 indoles,24 organometallic species,25 tryptamines,26 amines,27 allyl silanes28 and phenols.29 An interesting cyclization was observed with different internal alkynes and PhNHSCF3 furnishing the corresponding trifluoromethylthio substituted indoles,30 benzofurans and benzothiophenes,31 1H-isochromen-1-ones,32 2H-benzo[e][1,2]thiazine 1,1-dioxides33 and benzofulvenes.34 A recently developed trifluoromethanesulfonyl hypervalent iodonium ylide was shown to be an effective trifluoromethylthiolating agent after an in situ reduction of trifluoromethanesulfonyl group.35N-(Trifluoromethylthio)succinimide was utilized in a selective trifluoromethylthiolation of arenes,36 while an in situ-generated reagent from AgSCF3 and NCS was applied for the trifluoromethylthiolation of terminal alkynes.37N-Trifluoromethylthiophthalimide was employed in trifluoromethylthiolation of boronic acids,38 alkynes,39 amines and thiols,40 and a combination with cinchona alkaloids was utilized in a catalytic asymmetric trifluoromethylthiolation of oxindoles41 and β-ketoesters.42 One interesting trifluoromethylthiolating agent is a thioperoxide-based reagent containing a reactive O–S bond.43,44 It was found to be effective in trifluoromethylthiolation of boronic acids,45 Grignard reagents, alkynes, indoles, β-ketoesters, oxindoles, indoles,46 sodium sulfinates47 and carboxylic acids.48 It was also utilized in the activation of thioglycoside donors49 and in catalytic asymmetric trifluoromethylthiolations.50N-Trifluoromethylthiosaccharin51 was applied in the trifluoromethylthiolation of alcohols, amines, thiols, arenes,52 aldehydes, ketones, acyclic β-ketoesters, and alkynes. Functionalization of allylic alcohols furnished the corresponding trifluoromethyl sulfoxides via a [2,3]-sigmatropic rearrangement.53 A combination of AgSCF3 and trichloroisocyanuric acid was utilized as an in situ electrophilic SCF3 source in enantioselective catalytic functionalization of oxindoles54 and in the synthesis of 3-((trifluoromethyl)thio)-4H-chromen-4-ones.55 Carbonyl compounds56 and arenes57 were trifluoromethylthiolated with a new N-((trifluoromethyl)thio)benzenesulfonamide type of reagent.
Trifluoromethyl disulfides are important as precursors of biologically active trifluoromethyl thiosulfonates58,59 and can be prepared from thiols60 and CF3SCl or uncommon pyrole-SCF3 derivatives,61 upon reaction of bis(trifluoromethyl) trisulfide and organolithium reagents,62 and via the photochemical reaction of trifluoromethylated thioesters with disulfides.63 The first of these20 is not very convenient or safe, while the latter two methods62,63 were neither selective nor synthetically useful. The recently developed reagents N-trifluoromethylthiosaccharin51 and N-trifluoromethylthiophthalimide40 were demonstrated to be suitable for the synthesis of trifluoromethyl disulfides. We were interested in the reactivity of 1 with thiols because several different transformations are possible. Aromatic ring functionalization could take place; oxidation of thiols into the corresponding disulfides and synthesis of the trifluoromethyl disulfides are the other options. Here, we report on a straightforward, selective and efficient synthesis of trifluoromethyl disulfides.
Results and discussion
Initially, 4-methylthiophenol 2a was reacted with 1 in dichloromethane (DCM), and only traces of 4-methylphenyl trifluoromethyl disulfide 3a were noted (Table 1, entry 1). Yields were determined by 19F NMR spectroscopy using octafluoronaphthalene as internal standard. Structures of the products were further verified independently. It is known that the electrophilic power of 1 is not high enough to react without promoters/additives.23 Several potential additives in the transformation of 2a were examined; the results are summarized in Table 1.|
|
||
|---|---|---|
| Entry | Additive/(equiv.) | Yieldb (%) |
| a Reaction conditions: 2a (0.2 mmol), 1 (0.24 mmol), additive, CH2Cl2 (2 mL), Ar atmosphere, rt, 12 h. b Yields were determined by 19F NMR spectroscopy using octafluoronaphthalene as internal standard. c Di(4-methylphenyl) disulfide was the main product. d Ratio 3a/the disulfide by-product (1.9/1) was determined by 1H NMR spectroscopy. e Ratio 3a/the disulfide by-product (3.5/1) was determined by 1H NMR spectroscopy. f Isolated yield. | ||
| 1 | — | Tracesc |
| 2 | (CF3CO)2O/2 | 51 |
| 3 | CF3COOH/2 | 35d |
| 4 | BF3·Et2O/5 | 46e |
| 5 | TMSOTf/2 | 57 |
| 6 | Tf2O/2 | 51 |
| 7 | CF3SO3H/0.5 | 20 |
| 8 | CF3SO3H/1.0 | 62 |
| 9 | CF3SO3H/1.2 | 81 [72]f |
| 10 | CH3SO3H/0.5 | 9 |
| 11 | CH3SO3H/1.0 | 38 |
| 12 | CH3SO3H/1.3 | 82 [72]f |
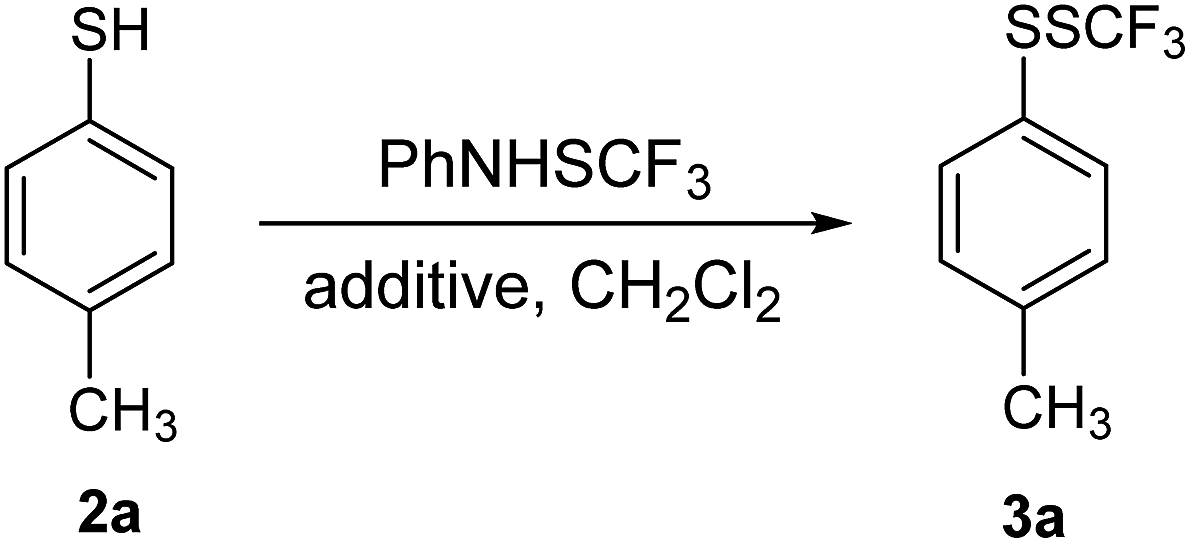
Reaction of 2a with 1 in the presence of 2 equivalents of trifluoroacetic anhydride (TFAA) gave 3a in 51% yield (entry 2). Transformations of 2a in the presence of some other Lewis acids were not very selective and efficient (entries 3–6). Consequently, we turned our attention to Brønsted acids such as trifluoromethanesulfonic acid (TfOH). Conversion of 2a in the presence of 0.5 equivalents of TfOH was only 20% (entry 7). The reaction progressed well in the presence of 1 equivalent of TfOH, however 10% of 2a remained unreacted (entry 8). Full conversion of 2a was achieved with 1.2 equivalents of TfOH, and 3a was isolated in good yield (entry 8); while a little amount of 2a remained unreacted with 1.1 equivalents of TfOH. Methanesulfonic acid (MSA) was tested similarly. Conversion of 2a in the presence of 0.5 equivalents of MSA was as low as 9% (entry 10), while it rose up to 38% when using 1 equivalent of MSA (entry 11). Transformation of 2a in the presence of 1.2 equivalents of MSA was almost complete, while full conversion was achieved in the presence of 1.3 equivalents of MSA (entry 12). The product 3a was isolated in a good yield as a sole product. The best reaction selectivity was obtained in entries 9 and 12 yielding 3a only. We decided to use MSA as an additive; however, in some cases better results were obtained with TfOH.
The role of solvent polarity on the reaction course of 2a with 1 was also examined (Table 2). Functionalization in hexane was completely selective; however, the conversion was not complete (entry 1). Selectivity in toluene was somewhat lower as well as the yield of 3a (entry 2). Diethyl ether would not be a suitable solvent because of low conversion of 2a into 3a (entry 3). Conversion in acetone was good, but some 2a remained unreacted (entry 4). Methanol was a poor solvent for this transformation, since the conversion of 2a remained low (entry 5). In acetonitrile, reaction took place well; however, a part of 2a remained unreacted (entry 6). Functionalization was completely suppressed in water where only traces of 3a were detected, and 2a and 1 were recovered (entry 7). Interestingly, good yields were obtained in hexane and in acetonitrile in spite of a remarkably different polarity. The protic solvents and water are obviously not suitable for this transformation. Dichloromethane was found to be the best solvent for this reaction (entry 8).
|
|
||
|---|---|---|
| Entry | Solvent | Yieldb (%) |
| a Reaction conditions: 2a (0.2 mmol), 1 (0.24 mmol), MSA (0.26 mmol), solvent (2 mL), Ar atmosphere, rt, 12 h. b Yields were determined by 19F NMR spectroscopy using octafluoronaphthalene as internal standard. c Isolated yield. | ||
| 1 | Hexane | 71 |
| 2 | Toluene | 70 |
| 3 | Diethyl ether | 36 |
| 4 | Acetone | 63 |
| 5 | Methanol | 32 |
| 6 | Acetonitrile | 62 |
| 7 | Water | Traces |
| 8 | Dichloromethane | 82 [72]c |
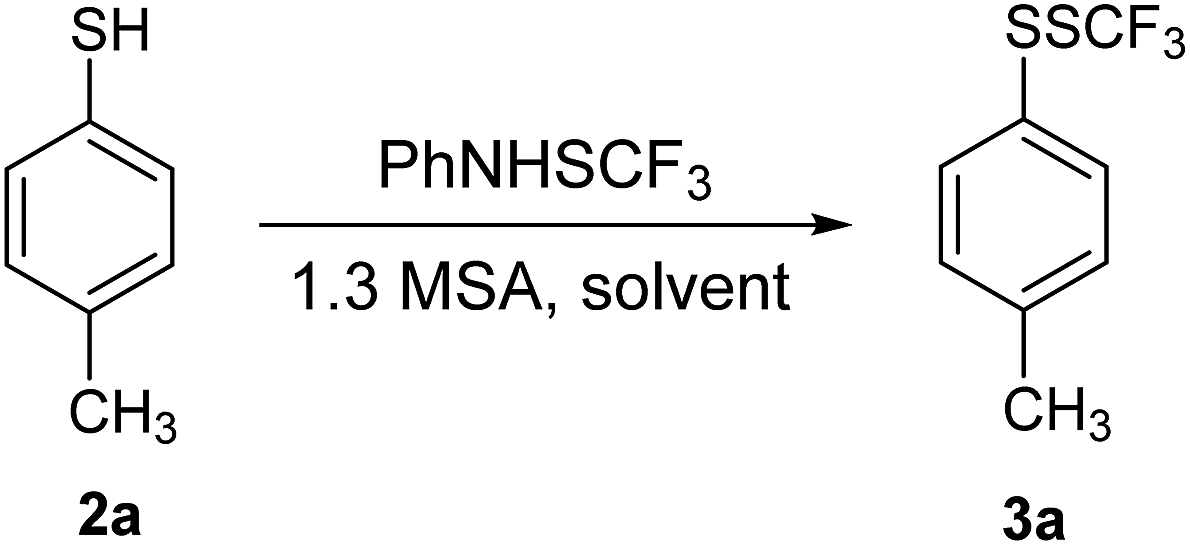
The reactivity of different thiols with 1 in dichloromethane under argon atmosphere was tested, and the optimal amount of acid (1.2 and 1.3 equivalents) was used. The results are summarized in Table 3. The model thiol 2a was efficiently converted into 3a, which was isolated on 0.5 mmol scale in higher yield than on a 0.2 mmol scale (entry 1, Table 3).
|
|
||||
|---|---|---|---|---|
| Entry | Ar | Acid/equiv. | 3 | Yieldb (%) |
| a Reaction conditions: (a) Thiols 2a–2j (0.5 mmol), 1 (0.6 mmol) and MSA (0.6–0.7 mmol) in DCM (5 mL) at rt for 12 h under Ar. Thiols (2k–2s) were functionalized under the same reaction conditions in the presence of TfOH (0.6 mmol). b Isolated yields. | ||||
| 1 | 4-Me–C6H4– 2a | MSA/1.3 | 3a | 82 |
| 2 | 4-OMe–C6H4– 2b | MSA/1.2 | 3b | 76 |
| 3 | 4-OH–C6H4– 2c | MSA/1.3 | 3c | 88 |
| 4 | 2-OMe–C6H4– 2d | MSA/1.2 | 3d | 91 |
| 5 | 2,4-DiMe–C6H3– 2e | MSA/1.2 | 3e | 90 |
| 6 | 2,5-DiMe–C6H3– 2f | MSA/1.3 | 3f | 92 |
| 7 | 3,5-DiMe–C6H3– 2g | MSA/1.3 | 3g | 89 |
| 8 | 4-iPr–C6H4– 2h | MSA/1.4 | 3h | 90 |
| 9 | 2-Naphthyl– 2i | MSA/1.3 | 3i | 91 |
| 10 | 4-Cl–C6H4– 2j | MSA/1.3 | 3j | 84 |
| 11 | 4-H–C6H4– 2k | TfOH/1.2 | 3k | 75 |
| 12 | 3-OMe–C6H4– 2l | TfOH/1.2 | 3l | 88 |
| 13 | 4-F–C6H4– 2m | TfOH/1.2 | 3m | 83 |
| 14 | 2-F–C6H4– 2n | TfOH/1.2 | 3n | 83 |
| 15 | 2,4-DiF–C6H3– 2o | TfOH/1.2 | 3o | 80 |
| 16 | 3,4-DiCl–C6H3– 2p | TfOH/1.2 | 3p | 88 |
| 17 | 2,5-DiCl–C6H3– 2q | TfOH/1.2 | 3q | 89 |
| 18 | 3-CF3–C6H4– 2r | TfOH/1.2 | 3r | 82 |
| 19 | 4-NO2–C6H4– 2s | TfOH/1.2 | 3s | 87 |
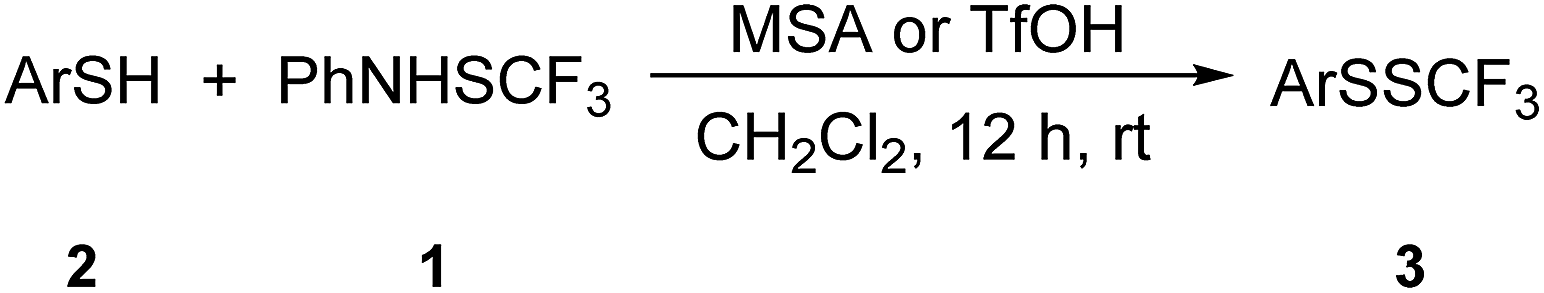
The highly activated 4-methoxythiophenol 2b selectively yielded 3b in the presence of MSA, whereas TfOH produced poorer results. A small amount of by-product was formed, which was not isolated, but, as could be judged from the NMR spectra, trifluoromethylthiolation of the aromatic ring took place. The functionalization of 4-hydroxythiophenol 2c, 2-methoxythiophenol 2d and 2,4-dimethylthiophenol 2e smoothly yielded the corresponding trifluoromethyl disulfides 3c, 3d and 3e in the presence of MSA (entries 3–5, Table 3). The less electron-rich 2,5-dimethylthiophenol 2f, 3,5-dimethylthiophenol 2g, 4-i-propylthiophenol 2h, 2-naphthalenethiol 2i and 4-chlorothiophenol 2j were selectively converted into their trifluoromethylthio derivatives 3f–3j in the presence of MSA (entries 6–10, Table 3). It was established that thiols bearing electron-withdrawing groups as a rule required stronger activation in comparison with the thiols bearing electron-donating groups. Thiophenol 2k, 3-methoxythiophenol 2l, fluoro-substituted thiophenols 2m–2o and dichloro-substituted thiophenols 2p and 2q were transformed into the desired trifluoromethyl disulfides 3k–3q in the presence of TfOH (entries 11–17, Table 3).
The reactions were selective without substantial formation of other side-products. Functionalization of electron-deficient 3-(trifluoromethyl)thiophenol 2r and 4-nitrothiophenol 2s with 1 took place efficiently in the presence of TfOH yielding the desired products 3r and 3s (entries 18 and 19, Table 2). For illustration, 2p and 2s did not react completely in the presence of 1.5 equiv. of MSA, while the full conversion was observed in the presence of 1.2 equiv. of TfOH.
Reactivity of benzyl thiols with 1 was also examined (Table 4). 4-Methoxybenzyl thiol 4a was selectively transformed into its trifluoromethyl disulfide 5a in the presence of MSA (entry 1, Table 4). It was established that benzyl thiols were of higher reactivity than thiophenols and MSA was found to be a suitable additive regardless on the substituents. Reactions of benzyl thiols in the presence of TfOH were much more complex with more unidentified by-products. Benzyl thiol 4b, 4-fluorobenzyl thiol 4c, 4-chlorobenzyl thiol 4d, and 3-(trifluoromethyl)benzyl thiol 4e were efficiently converted into the corresponding trifluoromethyl disulfides 5b–5e (entries 2–5, Table 4). It is once again obvious, that a relationship for the higher yield exists between the nucleophilicity of the thiols and strength of the promoter, i.e. strongly nucleophilic thiols require milder activation. 1-Phenylethanethiol 4f was detected in a peel oil extract of Pontianak oranges as a mixture of enantiomers ((R)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(S)) = 76:24, and it contributes to the characteristic odor of this citrus fruit. The compound has a very low odour threshold of 0.005 ng L−1 in the air.64 Functionalization of racemic 4f with 1 in the presence of MSA led to the desired product 5f (entry 6, Table 4).
:(S)) = 76:24, and it contributes to the characteristic odor of this citrus fruit. The compound has a very low odour threshold of 0.005 ng L−1 in the air.64 Functionalization of racemic 4f with 1 in the presence of MSA led to the desired product 5f (entry 6, Table 4).
|
|
||||
|---|---|---|---|---|
| Entry | R | 5 | Yieldb (%) | |
| a Reaction conditions: Thiol 4 (0.5 mmol), 1 (0.6 mmol), MSA (0.6–0.65 mmol) in DCM (5 mL) at rt for 12 h under Ar. Entry 10: 4j (0.5 mmol), 1 (1.2 mmol), MSA (1.3 mmol) in DCM (5 mL) at rt for 12 h under Ar. b Isolated yields. | ||||
| 1 | 4-MeO–C6H4–CH2– | 4a | 5a | 94 |
| 2 | 4-H–C6H4–CH2– | 4b | 5b | 80 |
| 3 | 4-F–C6H4–CH2– | 4c | 5c | 78 |
| 4 | 4-Cl–C6H4–CH2– | 4d | 5d | 87 |
| 5 | 3-CF3–C6H4–CH2– | 4e | 5e | 93 |
| 6 | Ph(Me)CH– | 4f | 5f | 58 |
| 7 | n-C8H17– | 4g | 5g | 83 |
| 8 | n-C12H25– | 4h | 5h | 86 |
| 9 | c-C6H11– | 4i | 5i | 57 |
| 10 | –(CH2)6– | 4j | 5j | 88 |
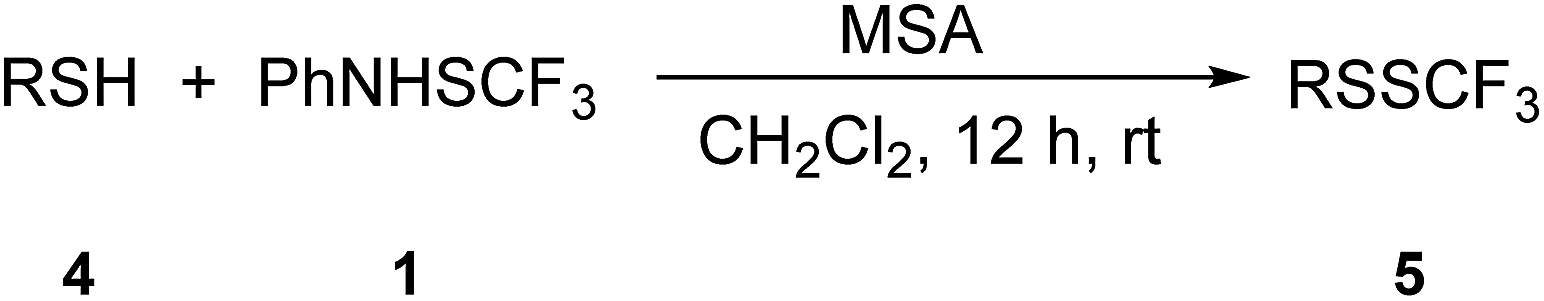
Some aliphatic thiols were also examined in reaction with 1. As could have been anticipated, the suitable additive was MSA. 1-Octanethiol 4g and 1-dodecanethiol 4h selectively furnished the related trifluoromethyl disulfides 5g and 5h (entries 7 and 8, Table 4). The reaction of cyclohexanethiol 4i yielded the corresponding product 5i in a reasonable yield (entry 9, Table 4). 1,6-Hexanedithiol 4j was tested, and double functionalization took place, thus yielding 5j in a high yield (entry 10, Table 4).
Furthermore, we focused on the reactivity of sterically hindered- and heteroaromatic thiols (Table 5). 2,4,6-Trimethylthiophenol 6a was successfully converted into trimethyl disulfide 7a in the presence of TfOH, while MSA was not an effective promoter. A remarkably sterically hindered thiol group in triphenylmethanethiol 6b was efficiently functionalized in the presence of TfOH, and product 7b was isolated in a high yield. 2-Phenylthiophenol 6c gave the expected trifluoromethyl disulfide 7c in the presence of 5 equiv. of p-toluenesulfonic acid hydrate. 4-Methyl-1,2-benzenedithiol 6d possesses two adjacent thiol groups, and both were functionalized with 1 in the presence of TfOH giving 7d.
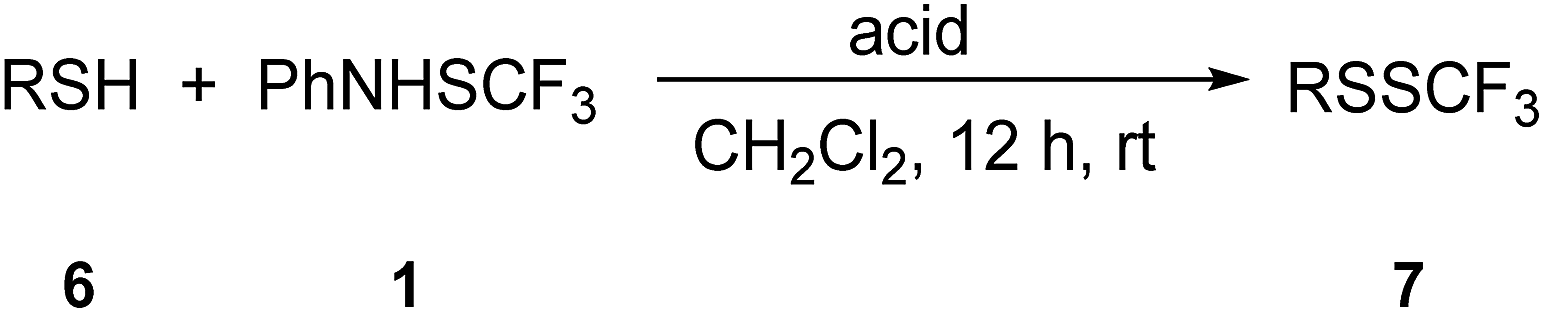
The transformation of 2-thiophenethiol 6e into 7e occurred in the presence of MSA, while selectivity dropped remarkably in the presence of TfOH. 2-Mercaptopyrimidine 6f and 2-mercaptobenzothiazole 6g were converted into the related trifluoromethyl disulfides 7f and 7g in the presence of 5 equiv. of p-toluenesulfonic acid hydrate. It could be concluded that sterically hindered- and heteroaromatic thiols could be effectively functionalized with 1.
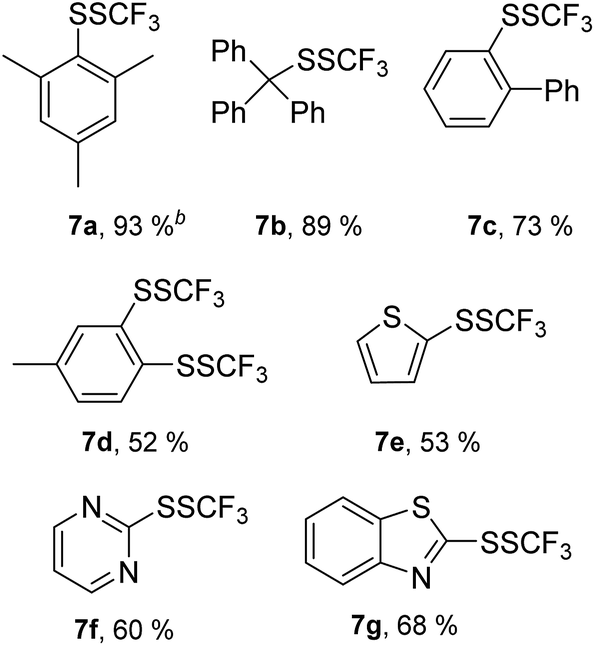
Next, we turned our attention to the structurally diverse, acid-sensitive and biologically important thiols (Table 6). It is known that the camphor skeleton could undergo an acid-catalyzed rearrangement, and it is frequently used in organocatalysis as well as a chiral auxiliary. (±)-7,7-Dimethylbicyclo-1-(mercaptomethyl)bicyclo[2.2.1]heptan-2-one 8a yielded the expected trifluoromethyl disulfide 9a in the presence of TfOH, and no rearrangement was noted.

This is a significant indication that the applied acidic reaction conditions turned to be compatible with acid-labile substrates. 7-Mercapto-4-methylcoumarin 8b was selectively converted into 3j in the presence of TfOH in spite of the presence of the acid-labile lactone group. Amide 8c was converted into 9c in the presence of TfOH in a moderate yield. The functionalization of 2-mercapto-1-phenylethanone 8d with 1 in the presence of BF3·Et2O led to the related α-trifluoromethyldisulfido ketone 9d. The reaction of thiobenzoic acid 8e in the presence of TfOH produced 9e, although it was isolated in a moderate yield.
In addition, we examined the reactivity of two biologically relevant thiols, i.e. the protected cysteine derivative 8f and 1-thioglucose derivative 8g. The both substrates are particularly challenging because of the acidic reaction conditions. The functionalization of 8f proceeded completely in the presence of 1.2 equiv. of MSA, and 9f was obtained in a 58% yield. The reaction of 8g in the presence of 1.2 equiv. of MSA took place smoothly; however, two sets of signals were observed in 1H NMR spectrum. Interestingly, the transformation of 8g in the presence of 1.2 equivalents of TfOH furnished a single stereoisomer 9g, and no epimerization took place. We were pleased that protecting groups were compatible with the acidic reaction conditions and that epimerization in the case of 8g could be avoided.
There is hardly any mention of trifluoromethyl thiosulfonates in the literature.65 Recently, an efficient synthetic method starting from sodium sulfonates was published.47 Thiosulfonates possess different biological activities58,59 and could be utilized as sulfenylating agents.66 Upon reaction with 1 in the presence of 5 equiv. of pTSA·H2O, sodium sulfinates 10a and 10b produced the corresponding trifluoromethyl thiosulfonates 11a and 11b in high yields (Scheme 1).
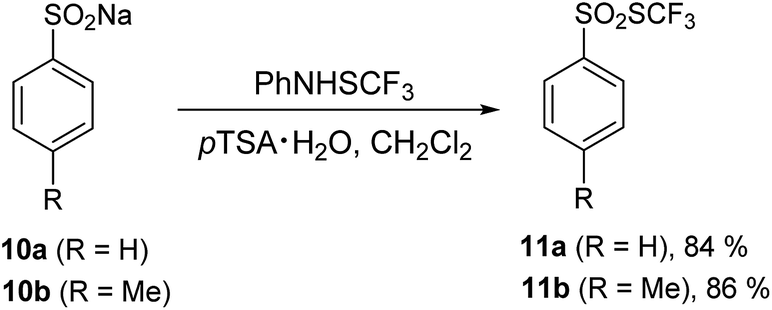 | ||
| Scheme 1 Transformation of sodium sulfinates with PhNHSCF3. | ||
Additionally, the reactivity of the selenium analog 12 was examined (Scheme 2). Benzeneselenol 12 reacted fully with 1 in the presence of 1.5 equivalents of MSA, giving product 13 and a substantial amount of diphenyl diselenide.
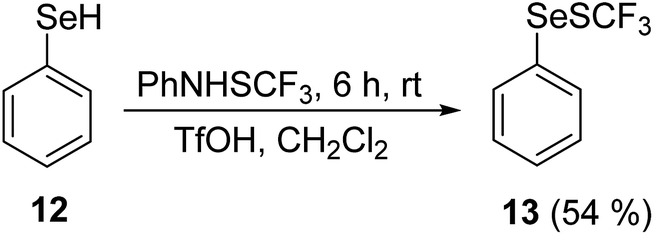 | ||
| Scheme 2 Reaction of benzeneselenol 12 with PhNHSCF3. | ||
The reaction of 12 in the presence of 1.2 equiv. of TfOH was considerably more selective, and phenylselenyl(trifluoromethyl) sulfide 13 was obtained as the sole product. The starting material 12 already contained a small amount of diphenyl diselenide; however, no appreciable additional oxidation of 12 took place.
The SSCF3 group is a relatively strong electron-withdrawing group, and the reactivity of trifluoromethyl disulfides is substantially unexplored. We were interested in how the SSCF3 group affects the reactivity of the aromatic ring. A model disulfide 3a was reacted with a mixture of concentrated HNO3/H2SO4 at 50 °C (Scheme 3).
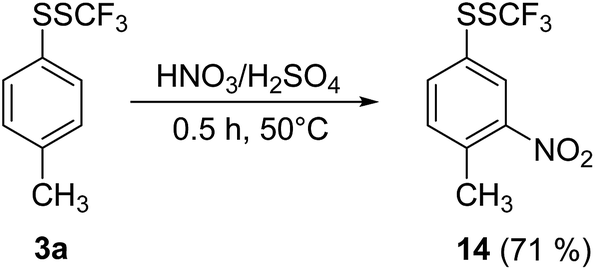 | ||
| Scheme 3 Functionalization of 3a with HNO3/H2SO4. | ||
Trace amounts of the 3-nitro regioisomer were also detected in the crude reaction mixture. No oxidation of the sulfur atoms or of the methyl group was noted, while nitration took place, furnishing 2-nitro derivative 14 as the main product.
Mechanism
A detailed reaction mechanism is not known, although several conclusions could be drawn. The relative reactivity of the aryl-substituted thiols with 1 was determined, and a slope of the Hammett linear free energy relationship was obtained to be ρ = −1.65 with a good correlation (r2 > 0.95) (Fig. 1).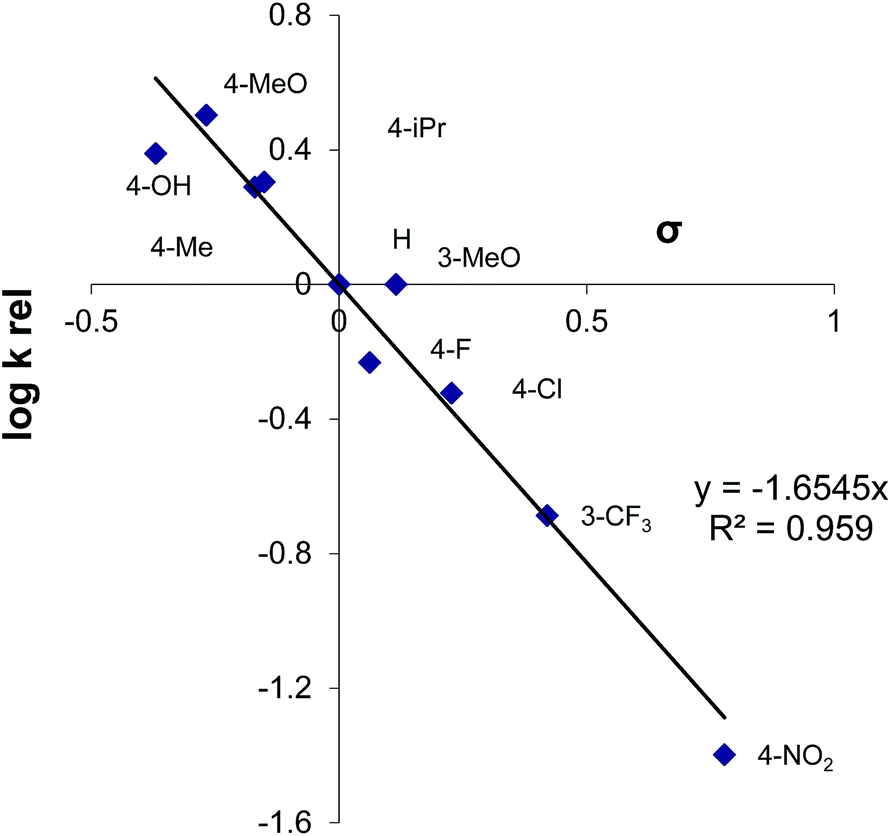 | ||
| Fig. 1 Hammett correlation for functionalization of the aryl-substituted thiols with 1. | ||
The more electron-deficient thiols are of lower reactivity than the electron-rich ones, thus indicating that the electron density on the sulfur atom is more important than the acidity of the starting thiol. The Hammett correlation indicated that an important amount of the positive charge was developed in the transition state. The formation of the thiolate anion under the acidic reaction conditions was likely not to be a significant process, but thiols remained in the molecular form that is presumably able to react with the activated form of the reagent 1. An unactivated reagent 1 is essentially an amine of relatively low electrophilic power. The acid seemingly protonates 1 on the nitrogen atom, thus generating a species of higher electrophilicity, able to react with thiols as nucleophiles. The reaction of 2a with 1/TfOH was examined in the presence of TEMPO, and the full conversion of 2a into 3a was noted. The yield of the product was practically the same as without TEMPO, and radicals are not likely to be important reaction intermediates.9e
Some additional experiments were performed in order to get a deeper insight into a reaction mechanism. The acid has a double role in this transformation; it acts as a promoter and as a reagent, since one of the final products is a salt of an aromatic amine. Reagent 1 was dissolved in CDCl3 and separately treated with MSA and TfOH in two NMR tubes. 1 reacted fully with TfOH immediately, and an instantaneous downfield shift of one major and two minor singlets appeared in the 19F NMR spectrum. This is an indication that protonation of 1 occurred, moreover it appears that there was an equilibrium between several species. On the contrary, reaction of 1 and MSA was far from completion after five minutes at room temperature; however there appeared three downfield shifted singlets in the 19F NMR spectrum, indicating a partial protonation of 1. MSA is likely too weak to protonate 1 in a weakly polar medium to an appreciable extent. The subsequent addition of 2a to both NMR tubes resulted in an immediate formation of 3a in both cases. In the case of TfOH, the activated form of 1 was already present, while addition of 2a in the case of MSA was a driving force to shift equilibrium with the activated form of 1 completely in the direction of formation of 3a. Based on the Hammett ρ value, we proposed a reaction of the protonated 1 with thiol as a nucleophilic substitution on the sulfur atom in a bimolecular reaction.
To elucidate further the reaction course, the quantum chemical calculations were performed. 1, methanethiol and methanesulfonic acid were chosen as a model system. Since the formation of ionic intermediates and/or transition states was proposed, calculations in vacuum would give erroneous results, therefore all calculations were carried out for species in dichloromethane solution, using the Poisson–Boltzmann model. Still, when energies of the participating species were computed individually, some computed energies were unrealistically high. In poorly polar medium, such as dichloromethane, extensive ion pairing exists, and when this was taken into account, a more realistic picture was obtained, presented in Fig. 2.
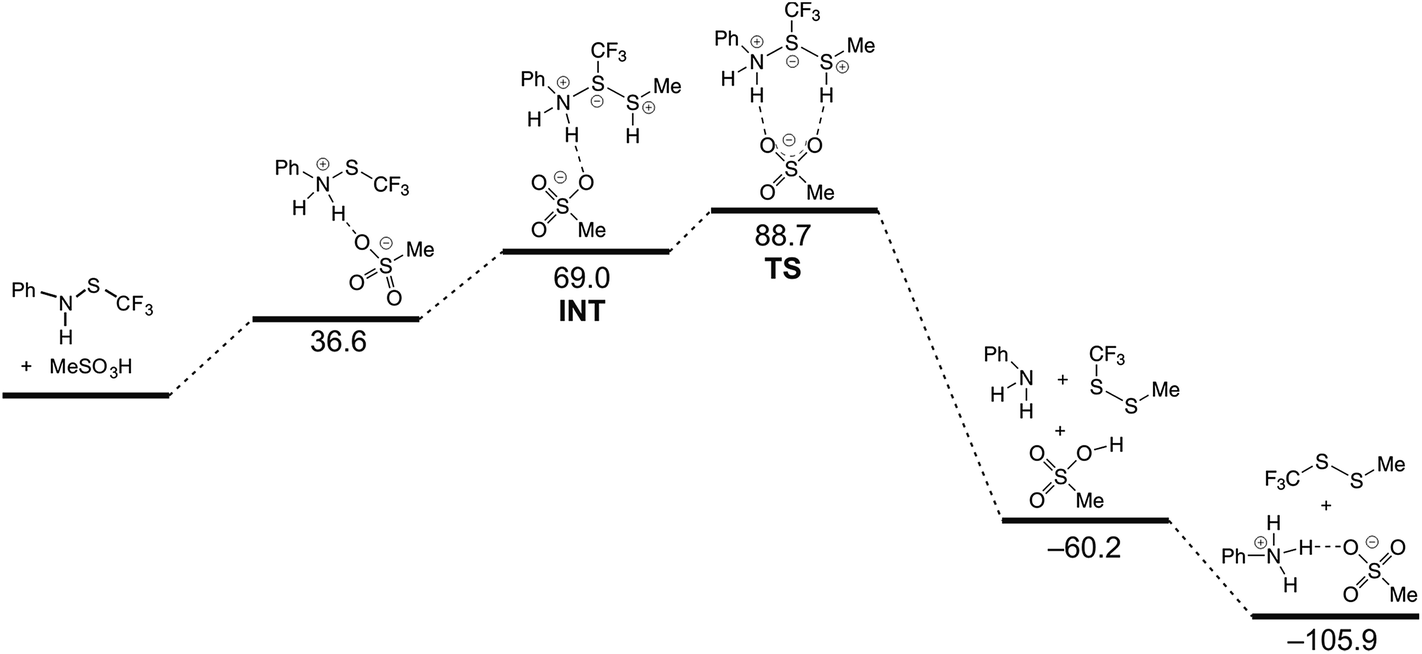 | ||
| Fig. 2 Energy diagram of the reaction of N-[(trifluoromethyl)thio]aniline with methanethiol and methanesulfonic acid in dichloromethane, computed at MPW1K/6-311+G** level. Enthalpies in kJ mol−1, in first two structures methanethiol is omitted. | ||
The reaction is, according to calculations, a two-step (A + D) nucleophilic substitution, starting with attack of thiol to a protonated amine. In the first step of the reaction, an intermediate is formed in a shallow depression. In this step, no appreciable activation barrier was found. The central S atom in the intermediate bears formally ten valence electrons, which causes the N–S–S bonding to be essentially a 3c-4e (hypervalent) bond. This is reflected in elongated bonds, nearly linear arrangement of N–S–S atoms and typical charge distribution (see ESI†). In the intermediate and the transition state a charge of +0.561 and +0.775 a.u. (NBO), respectively, appears on methanethiol moiety, which is in accordance with value of the measured Hammett ρ constant. The process in and around the transition state is essentially a movement of sulfonate anion, attached initially to an N–H proton, towards the S–H proton. Removal of the latter proton causes simultaneous cleavage of the NH–O and N–S bonds and disintegration of the aggregate into products (Fig. 3).
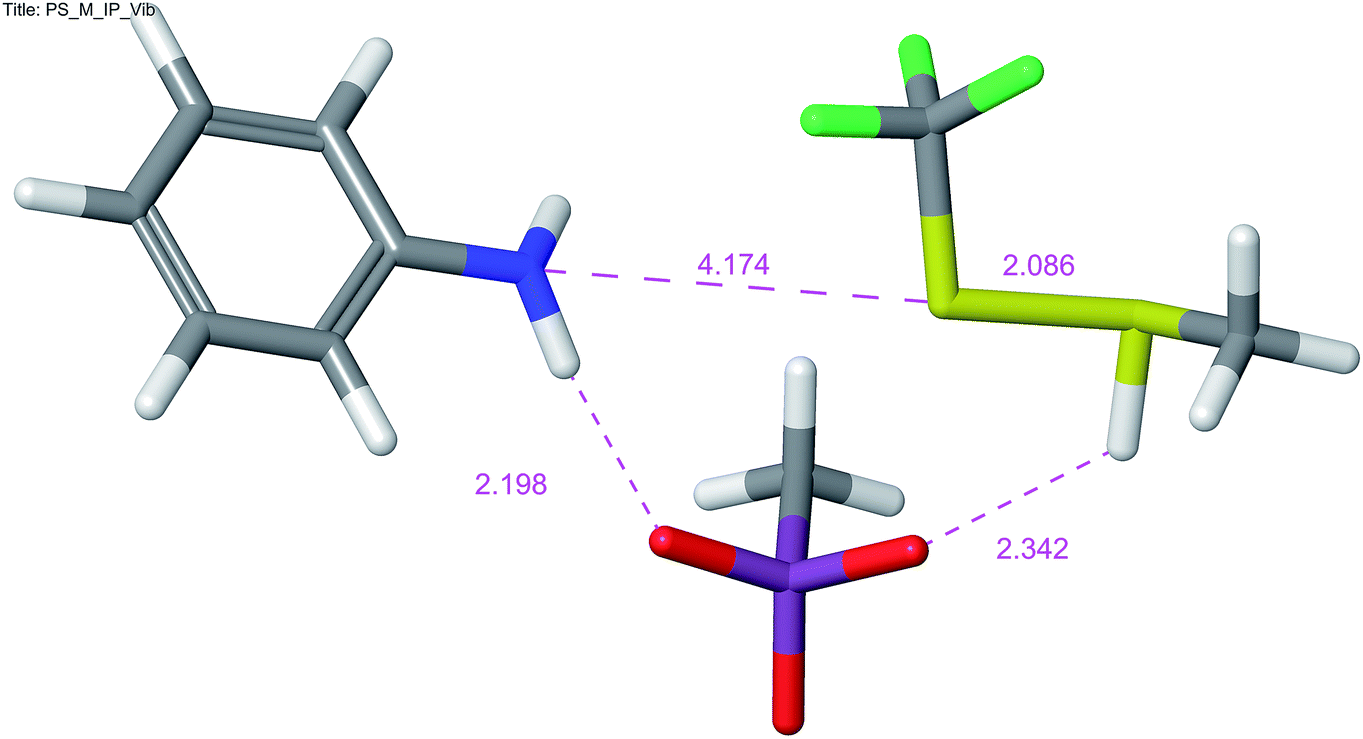 | ||
| Fig. 3 Transition state for the nucleophilic substitution on the sulfur atom of 1 with methanethiol, MPW1K/6-311+G**. | ||
Conclusions
The reactivity of the electrophilic trifluoromethylthiolating reagent PhNHSCF3 was tested on aryl-, benzyl-, alkyl-, heteroaryl-, sterically hindered-, biologically relevant- and acid sensitive thiols in the presence of acidic additives. The aryl-substituted thiols reacted well, regardless of the nature of the substituents. Trifluoromethyl disulfides were obtained as the sole products, no aromatic ring functionalization or parent disulfides formation were noted. Some acid-sensitive substrates were efficiently functionalized, thus proving compatibility with the acidic reaction conditions. The more electron-rich thiophenols required milder activation (methanesulfonic acid), and the more electron-deficient thiols required triflic acid. The nucleophilicity of the starting thiols appears to be of higher importance than their acidity. Sodium sulfonates were demonstrated to be suitable substrates for the synthesis of trifluoromethyl thiosulfonates using 1 and p-toluenesulfonic acid hydrate as an additive. Benzeneselenol was conveniently transformed into PhSeSCF3, a novel type of selenium compounds. The reactivity of trifluoromethyl disulfide product 3a with HNO3/H2SO4 was examined. Nitration of the aromatic ring took place, and no oxidation of sulfur atoms or methyl group was observed. The Hammett correlation analysis on the functionalization of the substituted thiophenols revealed the formation of the electron-deficient intermediate ρ = −1.65. MSA or TfOH most likely protonated 1 and enhanced its reactivity due to the stronger polarization between sulfur and nitrogen atoms. The generated sulfur electrophile is then able to react with thiols. Reactions in the presence of free radical TEMPO revealed that radicals are not very likely to be important reaction intermediates. The quantum chemical calculations indicated protonation of 1 in the initial stage, followed by a nucleophilic attack of thiol to the protonated 1, forming an intermediate, which decomposes through a transition state in which the sulfonate anion, attached initially by hydrogen bonding to an N–H proton, approaches the S–H proton. Removal of the S–H proton brings about an instantaneous collapse of the transition aggregate into products. In the intermediate and transition state a considerably positive charge is formed on the thiol moiety, which is in a good agreement with the Hammett correlation study.Experimental section
General information
All reactions were carried under an argon atmosphere with stirring at room or elevated temperature. Dichloromethane (>99.9%) was used as received. Most of thiols, benzeneselenol, other catalysts and acids were obtained from commercial sources and used as received.Crude trifluoromethylthiolated products were purified by column chromatography on silica gel (63–200 μm, 70–230 mesh ASTM; Fluka) using hexane or hexane/diethyl ether. TLC was performed on Merck-60-F254 plates using mixtures of hexane and diethyl ether. The melting points were determined in open-capillaries on Büchi 535 apparatus and are uncorrected. All products were characterized by their 1H NMR, 19F NMR, 13C NMR spectra, IR, HRMS and/or elemental analysis. HRMS data were obtained on Agilent 6224 Accurate Mass TOF LC/MS instrument (ESI-TOF) at Infrastructure Centre at UL FCCT in Ljubljana and on Thermo Scientific Q-Exactive spectrometer with Ion Max ion source equipped with a Syagen Technology PhotoMate Krypton lamp (APCI and APPI with orbitrap mass analyzer) in the Central Laboratory for Environmental, Plant & Microbial Metabolomics at the Karl-Franzens-University in Graz. The 1H and 13C NMR spectra were recorded on Bruker Avance 300 DPX, Bruker Avance III 500, and on Varian System 600 MHz instruments. The 19F NMR spectra were only recorded on Bruker Avance III 500 instrument. Chemical shifts are reported in δ (ppm) values relative to the TMS (δ = 0.00 ppm) and to the residual CHCl3 (δ = 7.26 ppm) for 1H NMR, to the central line of CDCl3 (δ = 77.0 ppm) and to the central line of acetone-d6 (δ = 30.83 ppm) for 13C NMR. 19F NMR spectra are referred to CFCl3 (δ = 0.00 ppm).
Representative procedure for the trifluoromethylthiolation of thiols with PhNHSCF3
To a solution of 4-methybenzenethiol 2a (0.5 mmol, 62 mg) in dichloromethane (5 mL), N-[(trifluoromethyl)thio]aniline 1 (0.6 mmol, 116 mg) and trifluoromethanesulfonic acid (0.65 mmol, 63 mg) were added and the mixture was stirred under argon for 12 h. The mixture was diluted with 15 mL of CH2Cl2, washed with aqueous solution of NaHCO3, water and dried over anhydrous Na2SO4. After filtration, the solvent was evaporated and the crude reaction mixture was analyzed by 1H and 19F NMR spectroscopy. Pure product 3a as yellow oil (92 mg, 82%) was obtained after column chromatography on silica gel using hexane as eluent.The same procedure was used also in the case of benzeneselenol 12.
Experimental procedure for nitration of 3a
65% nitric acid (3.1 mmol, 300 mg) and 98% sulfuric acid (1.5 mmol, 150 mg) were added to 3a (0.6 mmol, 135 mg), and the mixture was stirred at 50 °C for 30 minutes. Full consumption of 3a was noted by TLC. The reaction mixture was cooled to rt, diluted with 15 mL of dichloromethane washed with aqueous solution of NaHCO3, water and dried over anhydrous Na2SO4. After filtration, the solvent was evaporated and the crude reaction mixture was analyzed by 1H and 19F NMR spectroscopy. Pure product 14 as yellow solid (114 mg, 71%) was obtained after column chromatography on silica gel using hexane/diethyl ether (1/10) as eluent.Determination of the relative reactivity of the substituted thiophenols in the Hammett correlation analysis
The relative rates of substituted thiophenols (Fig. 1) were determined by competitive reactions as follows: to a mixture of two substrates (reference PhSH (0.2 mmol) and the examined substituted thiophenol (0.2 mmol)), PhNHSCF3 (0.2 mmol) and MSA or TfOH (0.24–0.26 mmol) were added, and the mixture was stirred for 12 h at rt. The reaction was quenched with water, and products were extracted with DCM (2 × 10 mL) and washed with water. The phases were separated and the organic layer was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the reaction mixture was analyzed by 19F NMR. Octafluoronaphthalene was used as internal standard. The difference in reactivity of PhSH and 4-nitrothiophenol was too high to obtain reliable krel in this manner. For this reason, 4-chlorothiophenol was taken as a second reference molecule for 4-nitrothiophenol. The relative reactivity of 4-nitrothiophenol vs. PhSH was therefore determined indirectly: k(4-NO2)/k(4-H) was obtained from: k(4-Cl)/k(4-H) and k(4-Cl)/k(4-NO2). Relative reactivities expressed by the relative rate factors kR were calculated from the equation79kR = kA/kB = log((A − X)/A)/log((B − Y)/B), derived from the Ingold–Shaw relation,80 where A and B are the amounts of starting material and X and Y the amounts of products derived from them. The relative rate factors thus obtained, presented in Fig. 1, are the averages of at least three measurements.Acknowledgements
J. Robin and B. Alič for technical support, Prof. K. Francesconi and Dr K. B. Jensen at the Karl-Franzens-University in Graz, Dr D. Urankar and Prof. J. Košmrlj for HRMS, Mrs T. Stipanovič and Prof. B. Stanovnik for elemental combustion analyses, and the Ministry of Higher Education, Science and Technology (P1-0134) for financial support are gratefully acknowledged.References
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) Handbook of Fluorous Chemistry, ed. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (d) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (e) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (f) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef PubMed; (g) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (h) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (b) L. Hunter, Beilstein J. Org. Chem., 2010, 6, 38 Search PubMed; (c) G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed.
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (b) C. Dalvit and A. Vulpetti, ChemMedChem, 2012, 7, 262–272 CrossRef CAS PubMed.
- (a) H.-J. Schneider, Chem. Sci., 2012, 3, 1381–1394 RSC; (b) D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 2175–2186 RSC; (c) K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- (a) Z. Jin, G. B. Hammond and B. Xu, Aldrichimica Acta, 2012, 45, 67–83 CAS; (b) N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6, 65 CrossRef PubMed; (c) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed; (d) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed; (e) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (f) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048–5050 CrossRef CAS PubMed; (g) X.-F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 1744–1754 CrossRef CAS PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS PubMed; (c) J. Xu, X. Liu and Y. Fu, Tetrahedron Lett., 2014, 55, 585–594 CrossRef CAS PubMed; (d) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS PubMed; (e) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (f) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef CAS; (g) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CAS.
- L. M. Yagupolskii, N. V. Kondratenko and G. N. Timofeeva, J. Org. Chem. USSR, 1984, 20, 103–106 Search PubMed.
- (a) T. Umemoto and J. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156–2164 CrossRef CAS; (b) J.-J. Yang, R. L. Kirchmeier and J. M. Shreeve, J. Org. Chem., 1998, 63, 2656–2660 CrossRef CAS PubMed; (c) S. Noritake, N. Shibata, S. Nakamura, T. Toru and M. Shiro, Eur. J. Org. Chem., 2008, 3465–3468 CrossRef CAS PubMed; (d) A. Matsnev, S. Noritake, Y. Nomura, E. Tokunaga, S. Nakamura and N. Shibata, Angew. Chem., Int. Ed., 2010, 49, 572–576 CrossRef CAS PubMed; (e) P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579–2586 CrossRef CAS PubMed; (f) A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120–9123 CrossRef CAS PubMed; (g) T. Liu, X. Shao, Y. Wu and Q. Shen, Angew. Chem., Int. Ed., 2012, 51, 540–543 CrossRef CAS PubMed; (h) R. Shimizu, H. Egami, Y. Hamashima and M. Sodeoka, Angew. Chem., Int. Ed., 2012, 51, 4577–4580 CrossRef CAS PubMed; (i) Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221–8224 CrossRef CAS PubMed.
- See, for example: (a) C. Zhang, Org. Biomol. Chem., 2014, 12, 6580–6589 RSC; (b) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513–1522 CrossRef CAS PubMed; (c) T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2014, 20, 16830–16845 CrossRef CAS PubMed; (d) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed; (e) W. Kong, M. Casimiro, N. Fuentes, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 13086–13090 CrossRef CAS PubMed; (f) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- See, for example: (a) J. Xu, B. Xiao, C.-Q. Xie, D.-F. Luo, L. Liu and Y. Fu, Angew. Chem., Int. Ed., 2012, 51, 12551–12554 CrossRef CAS PubMed; (b) X. Mu, T. Wu, H.-y. Wang, Y.-l. Guo and G. Liu, J. Am. Chem. Soc., 2012, 134, 878–881 CrossRef CAS PubMed; (c) B. A. Khan, A. E. Buba and L. J. Gooßen, Chem.–Eur. J., 2012, 18, 1577–1581 CrossRef CAS PubMed; (d) A. Hafner and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 3713–3715 CrossRef CAS PubMed; (e) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262–7263 CrossRef CAS PubMed; (f) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439–10442 CrossRef CAS PubMed; (g) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119–6122 CrossRef CAS PubMed; (h) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed.
- See, for example: (a) G. Shi, C. Shao, S. Pan, J. Yu and Y. Zhang, Org. Lett., 2015, 17, 38–41 CrossRef CAS PubMed; (b) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed; (c) X.-Y. Jiang and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 14177–14180 CrossRef CAS PubMed; (d) Q. Lu, C. Liu, Z. Huang, Y. Ma, J. Zhang and A. Lei, Chem. Commun., 2014, 50, 14101–14104 RSC; (e) L. Zhang, Z. Li and Z.-Q. Liu, Org. Lett., 2014, 16, 3688–3691 CrossRef CAS PubMed; (f) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539–542 CrossRef CAS PubMed; (g) X. Wu, L. Chu and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 2198–2202 CrossRef CAS PubMed; (h) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS PubMed; (i) C. Zhang, Adv. Synth. Catal., 2014, 356, 2895–2906 CrossRef CAS PubMed.
- See, for example: (a) R. J. Lundgren and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 9322–9324 CrossRef CAS PubMed; (b) N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 2878–2879 CrossRef CAS PubMed; (c) T. S. N. Zhao and K. J. Szabó, Org. Lett., 2012, 14, 3966–3969 CrossRef CAS PubMed; (d) O. A. Tomashenko, E. C. Escudero-Adán, M. M. Belmonte and V. V. Grushin, Angew. Chem., Int. Ed., 2011, 50, 7655–7659 CrossRef CAS PubMed; (e) G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600–8601 CrossRef CAS PubMed; (f) N. D. Ball, J. B. Gary, Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 7577–7584 CrossRef CAS PubMed; (g) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679–1681 CrossRef CAS PubMed; (h) N. D. Litvinas, P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 536–539 CrossRef CAS PubMed; (i) H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793–3798 CrossRef CAS PubMed; (j) L. Zhu, S. Liu, J. T. Douglas and R. A. Altman, Chem.–Eur. J., 2013, 19, 12800–12805 CrossRef CAS PubMed; (k) P. Chen and G. Liu, Synthesis, 2013, 45, 2919–2939 CrossRef CAS; (l) Y. Ye and M. S. Sanford, Synlett, 2012, 23, 2000–2013 CrossRef; (m) M. Hu, C. Ni and J. Hu, J. Am. Chem. Soc., 2012, 134, 15257–15260 CrossRef CAS PubMed; (n) T. Liu and Q. Shen, Eur. J. Org. Chem., 2012, 6679–6687 CrossRef CAS PubMed; (o) J. M. Larsson, S. R. Pathipati and K. J. Szabó, J. Org. Chem., 2013, 78, 7330–7336 CrossRef CAS PubMed.
- G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941–951 CrossRef CAS.
- K. Fauster, C. Kreutz and R. Micura, Angew. Chem., Int. Ed., 2012, 51, 13080–13084 CrossRef CAS PubMed.
- (a) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312–7314 CrossRef CAS PubMed; (b) K. Zhang, J.-B. Liu and F.-L. Qing, Chem. Commun., 2014, 50, 14157–14160 RSC; (c) L. M. Yagupolskii, N. V. Kondratenko and V. P. Sambur, Synthesis, 1975, 721–723 CrossRef CAS; (d) M. Hu, J. Rong, W. Miao, C. Ni, Y. Han and J. Hu, Org. Lett., 2014, 16, 2030–2033 CrossRef CAS PubMed; (e) S.-G. Li and S. Z. Zard, Org. Lett., 2013, 15, 5898–5901 CrossRef CAS PubMed; (f) W. A. Sheppard, J. Org. Chem., 1964, 29, 895–898 CrossRef CAS; (g) D. J. Adams, A. Goddard, J. H. Clark and D. J. Macquarrie, Chem. Commun., 2000, 987–988 RSC; (h) C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183–185 CrossRef CAS PubMed; (i) C.-P. Zhang and D. A. Vicic, Chem.–Asian J., 2012, 7, 1756–1758 CrossRef CAS PubMed.
- V. N. Boiko, Beilstein J. Org. Chem., 2010, 6, 880–921 CrossRef PubMed.
- (a) K. Yamaguchi, K. Sakagami, Y. Miyamoto, X. Jin and N. Mizuno, Org. Biomol. Chem., 2014, 12, 9200–9206 RSC; (b) C. Chen, Y. Xie, L. Chu, R.-W. Wang, X. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492–2495 CrossRef CAS PubMed; (c) C. Pooput, M. Medebielle and W. R. Dolbier Jr, Org. Lett., 2004, 6, 301–303 CrossRef CAS PubMed; (d) N. Santschi and A. Togni, J. Org. Chem., 2011, 76, 4189–4193 CrossRef CAS PubMed; (e) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754–757 CrossRef CAS PubMed; (f) S. Capone, I. Kieltsch, O. Flögel, G. Lelais, A. Togni and D. Seebach, Helv. Chim. Acta, 2008, 91, 2035–2056 CrossRef CAS PubMed; (g) S. Large, N. Roques and B. R. Langlois, J. Org. Chem., 2000, 65, 8848–8856 CrossRef CAS PubMed; (h) T. Billard, S. Large and B. R. Langlois, Tetrahedron Lett., 1997, 38, 65–68 CrossRef CAS; (i) G. Blond, T. Billard and B. R. Langlois, Tetrahedron Lett., 2001, 42, 2473–2475 CrossRef CAS; (j) L. Zhai, Y. Li, J. Yin, K. Jin, R. Zhang, X. Fu and C. Duan, Tetrahedron, 2013, 69, 10262–10266 CrossRef CAS PubMed; (k) Y. Huang, X. He, X. Lin, M. Rong and Z. Weng, Org. Lett., 2014, 16, 3284–3287 CrossRef CAS PubMed; (l) V. N. Movchun, A. A. Kolomeitsev and Y. L. Yagupolskii, J. Fluorine Chem., 1995, 70, 255–257 CrossRef CAS; (m) W. Zhong and X. Liu, Tetrahedron Lett., 2014, 55, 4909–4911 CrossRef CAS PubMed; (n) G. Danoun, B. Bayarmagnai, M. F. Gruenberg and J. L. Goossen, Chem. Sci., 2014, 5, 1312–1316 RSC; (o) N. J. W. Straathof, B. J. P. Tegelbeckers, V. Hessel, X. Wang and T. Noël, Chem. Sci., 2014, 5, 4768–4773 RSC; (p) B. Bayarmagnai, C. Matheis, E. Risto and L. J. Goossen, Adv. Synth. Catal., 2014, 356, 2343–2348 CrossRef CAS PubMed.
- (a) M. Rueping, N. Tolstoluzhsky and P. Nikolaienko, Chem.–Eur. J., 2013, 19, 14043–14046 CrossRef CAS PubMed; (b) Z. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548–1552 CrossRef CAS PubMed; (c) J. H. Clark, C. W. Jones, A. P. Kybett, M. A. McClinton, J. M. Miller, D. Bishop and R. J. Blade, J. Fluorine Chem., 1990, 48, 249–253 CrossRef CAS; (d) D. J. Adams and J. H. Clark, J. Org. Chem., 2000, 65, 1456–1460 CrossRef CAS PubMed.
- L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef CAS PubMed.
- S. Munavalli, D. K. Rohrbaugh, D. I. Rossman and H. D. Durst, J. Fluorine Chem., 1999, 98, 3–9 CrossRef CAS.
- (a) A. Tlili and T. Billard, Angew. Chem., Int. Ed., 2013, 52, 6818–6819 CrossRef CAS PubMed; (b) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415–2428 CrossRef CAS PubMed; (c) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS PubMed; (d) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (e) C. Chen, L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 12454–12457 CrossRef CAS PubMed; (f) Q. Lefebvre, E. Fava, P. Nikolaienko and M. Rueping, Chem. Commun., 2014, 50, 6617–6619 RSC; (g) W. Yin, Z. Wang and Y. Huang, Adv. Synth. Catal., 2014, 356, 2998–3006 CrossRef CAS PubMed; (h) P. Nikolaienko, R. Pluta and M. Rueping, Chem.–Eur. J., 2014, 20, 9867–9870 CrossRef CAS PubMed; (i) C. Chen, X.-H. Xu, B. Yang and F.-L. Qing, Org. Lett., 2014, 16, 3372–3375 CrossRef CAS PubMed; (j) P. Zhu, X. He, X. Chen, Y. You, Y. Yuan and Z. Weng, Tetrahedron, 2014, 70, 672–677 CrossRef CAS PubMed; (k) D. Kong, Z. Jiang, S. Xin, Z. Bai, Y. Yuan and Z. Weng, Tetrahedron, 2013, 69, 6046–6050 CrossRef CAS PubMed; (l) Z. Wang, Q. Tu and Z. Weng, J. Organomet. Chem., 2014, 751, 830–834 CrossRef CAS PubMed; (m) Q. Xiao, J. Sheng, Q. Ding and J. Wu, Eur. J. Org. Chem., 2014, 217–221 CrossRef CAS PubMed; (n) F. Yin and X.-S. Wang, Org. Lett., 2014, 16, 1128–1131 CrossRef CAS PubMed; (o) X. Wang, Y. Zhou, G. Ji, G. Wu, M. Li, Y. Zhang and J. Wang, Eur. J. Org. Chem., 2014, 3093–3096 CrossRef CAS PubMed; (p) G. Landelle, A. Panossian, S. Pazenok, J.-P. Vors and F. R. Leroux, Beilstein J. Org. Chem., 2013, 9, 2476–2536 CrossRef PubMed; (q) J.-B. Liu, X.-H. Xu, Z.-H. Chen and F.-L. Qing, Angew. Chem., Int. Ed., 2015, 54, 897–900 CrossRef CAS PubMed; (r) X. Dai and D. Cahard, Synlett, 2015, 16, 40–44 Search PubMed.
- A. Ferry, T. Billard, B. R. Langlois and E. Bacqué, J. Org. Chem., 2008, 73, 9362–9365 CrossRef CAS PubMed.
- A. Ferry, T. Billard, B. R. Langlois and E. Bacqué, Angew. Chem., Int. Ed., 2009, 48, 8551–8555 CrossRef CAS PubMed.
- A. Ferry, T. Billard, E. Bacque and B. R. Langlois, J. Fluorine Chem., 2012, 134, 160–163 CrossRef CAS PubMed.
- F. Baert, J. Colomb and T. Billard, Angew. Chem., Int. Ed., 2012, 51, 10382–10385 CrossRef CAS PubMed.
- Y. Yang, X. Jiang and F.-L. Qing, J. Org. Chem., 2012, 77, 7538–7547 CrossRef CAS PubMed.
- S. Alazet, K. Ollivier and T. Billard, Beilstein J. Org. Chem., 2013, 9, 2354–2357 CrossRef PubMed.
- J. Liu, L. Chu and F.-L. Qing, Org. Lett., 2013, 15, 894–897 CrossRef CAS PubMed.
- M. Jereb and K. Gosak, Org. Biomol. Chem., 2015, 13, 3103–3115 CAS.
- J. Sheng, S. Li and J. Wu, Chem. Commun., 2014, 50, 578–580 RSC.
- J. Sheng, C. Fan and J. Wu, Chem. Commun., 2014, 50, 5494–5496 RSC.
- Y. Li, G. Li and Q. Ding, Eur. J. Org. Chem., 2014, 5017–5022 CrossRef CAS PubMed.
- Q. Xiao, J. Sheng, Z. Chen and J. Wu, Chem. Commun., 2013, 49, 8647–8649 RSC.
- Q. Xiao, H. Zhu, G. Li and Z. Chen, Adv. Synth. Catal., 2014, 356, 3809–3815 CrossRef CAS PubMed.
- (a) Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and M. Shibata, J. Am. Chem. Soc., 2013, 135, 8782–8785 CrossRef CAS PubMed; (b) S. Arimori, M. Takada and N. Shibata, Org. Lett., 2015, 17, 1063–1065 CrossRef CAS PubMed; (c) Z. Huang, Y.-D. Yang, E. Tokunaga and N. Shibata, Org. Lett., 2015, 17, 1094–1097 CrossRef CAS PubMed.
- C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046–2049 CrossRef CAS PubMed.
- S.-Q. Zhu, X.-H. Xu and F.-L. Qing, Eur. J. Org. Chem., 2014, 4453–4456 CrossRef CAS PubMed.
- K. Kang, C. Xu and Q. Shen, Org. Chem. Front., 2014, 1, 294–297 RSC.
- R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 1650–1653 CrossRef CAS PubMed.
- R. Pluta and M. Rueping, Chem.–Eur. J., 2014, 20, 17315–17318 CrossRef CAS PubMed.
- T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856–12859 CrossRef CAS PubMed.
- M. Rueping, X. Liu, T. Bootwicha, R. Pluta and C. Merkens, Chem. Commun., 2014, 50, 2508–2511 RSC.
- X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457–3460 CrossRef CAS PubMed.
- E. V. Vinogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 3125–3128 CrossRef CAS PubMed.
- X. Shao, T. Liu, L. Lu and Q. Shen, Org. Lett., 2014, 16, 4738–4741 CrossRef CAS PubMed.
- B. Ma, X. Shao and Q. Shen, J. Fluorine Chem., 2015, 171, 73–77 CrossRef CAS PubMed.
- X. Shao, C. Xu, L. Lu and Q. Shen, J. Org. Chem., 2015, 80, 3012–3021 CrossRef CAS PubMed.
- F. Hu, X. Shao, D. Zhu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 6105–6109 CrossRef CAS PubMed.
- H. He and X. Zhu, Org. Lett., 2014, 16, 3102–3105 CrossRef CAS PubMed.
- (a) X. Wang, T. Yang, X. Cheng and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 12860–12864 CrossRef CAS PubMed; (b) Q.-H. Deng, C. Rettenmeier, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2014, 20, 93–97 CrossRef CAS PubMed.
- C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316–9320 CrossRef CAS PubMed.
- Q. Wang, Z. Qi, F. Xie and X. Li, Adv. Synth. Catal., 2014, 357, 355–360 CrossRef PubMed.
- M. Maeno, N. Shibata and D. Cahard, Org. Lett., 2015, 17, 1990–1993 CrossRef CAS PubMed.
- X.-L. Zhu, J.-H. Xu, D.-J. Cheng, L.-J. Zhao, X.-Y. Liu and B. Tan, Org. Lett., 2014, 16, 2192–2195 CrossRef CAS PubMed.
- H. Xiang and C. Yang, Org. Lett., 2014, 16, 5686–5689 CrossRef CAS PubMed.
- S. Alazet, L. Zimmer and T. Billard, Chem.–Eur. J., 2014, 20, 8589–8593 CrossRef CAS PubMed.
- (a) S. Alazet and T. Billard, Synlett, 2015, 26, 76–78 CrossRef CAS; (b) S. Alazet, L. Zimmer and T. Billard, J. Fluorine Chem., 2015, 171, 78–81 CrossRef CAS PubMed.
- S. S. Block and J. P. Weidner, Nature, 1967, 214, 478–479 CrossRef CAS PubMed.
- J. P. Weidner and S. S. Block, J. Med. Chem., 1967, 10, 1167–1170 CrossRef CAS.
- S. Andreades, J. F. Harris Jr and W. A. Sheppard, J. Org. Chem., 1964, 29, 898–900 CrossRef CAS.
- D. M. Ceacareanu, M. R. C. Gerstenberger and A. Haas, Chem. Ber., 1983, 116, 3325–3331 CrossRef CAS PubMed.
- S. Munavalli, D. I. Rossman, D. K. Rohrbaugh, C. P. Ferguson and H. D. Banks, J. Fluorine Chem., 1993, 60, 85–91 CrossRef CAS.
- T. Billard, N. Roques and B. R. Langlois, J. Org. Chem., 1999, 64, 3813–3820 CrossRef CAS.
- A. Fischer, W. Grab and P. Schieberle, Eur. Food Res. Technol., 2008, 227, 735–744 CrossRef CAS.
- (a) J. P. Weidner and S. S. Block, J. Med. Chem., 1972, 15, 564–567 CrossRef CAS; (b) T. Billard, B. R. Langlois, S. Large, D. Anker, N. Roidot and P. Roure, J. Org. Chem., 1996, 61, 7545–7550 CrossRef CAS PubMed.
- M. G. Ranasinghe and P. L. Fuchs, J. Am. Chem. Soc., 1989, 111, 779–782 CrossRef CAS.
- (a) B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2005, 1, 415–432 CrossRef CAS; (c) D. Dolenc and B. Modec, New J. Chem., 2009, 33, 2344–2349 RSC.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001, http://www.chem.wisc.edu/~nbo5 Search PubMed.
- C. E. Aroyan, A. Dermenci and S. J. Miller, J. Org. Chem., 2010, 75, 5784–5796 CrossRef CAS PubMed.
- S. Pearson, W. Scarano and M. H. Stenzel, Chem. Commun., 2012, 48, 4695–4697 RSC.
- E. Juaristi and J. S. Cruz-Sánchez, J. Org. Chem., 1988, 53, 3334–3338 CrossRef CAS.
- J. Z. Chandanshive, B. F. Bonini, D. Gentili, M. Fochi, L. Bernardi and M. Comes Franchini, Eur. J. Org. Chem., 2010, 6440–6447 CrossRef CAS PubMed.
- S. Knoppe, N. Kothalawala, V. R. Jupally, A. Dass and T. Bürgi, Chem. Commun., 2012, 48, 4630–4632 RSC.
- S. J. Ratnakar, M. Woods, A. J. M. Lubag, Z. Kovács and A. D. Sherry, J. Am. Chem. Soc., 2008, 130, 6–7 CrossRef CAS PubMed.
- S. Bateja, S. Chandrashekhar, C. S. Bhandari and N. C. Sogani, J. Chin. Chem. Soc., 1979, 26, 173–176 CrossRef CAS PubMed.
- J. J. Garcia, B. E. Mann, H. Adams, N. A. Bailey and P. M. Maitlis, J. Am. Chem. Soc., 1995, 117, 2179–2186 CrossRef CAS.
- J. I. G. Cadogan, H. S. Hutchinson and H. McNab, J. Chem. Soc., Perkin Trans. 1, 1988, 2875–2879 RSC.
- M. Murár, G. Addová and A. Boháč, Beilstein J. Org. Chem., 2013, 9, 173–179 CrossRef PubMed.
- R. E. Pearson and J. C. Martin, J. Am. Chem. Soc., 1963, 85, 3142–3146 CrossRef CAS.
- C. K. Ingold and F. R. Shaw, J. Chem. Soc., 1927, 2918–2926 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 19F NMR spectra of all products, and computed geometries and energies of model species. See DOI: 10.1039/c5ra07316b |
| This journal is © The Royal Society of Chemistry 2015 |