
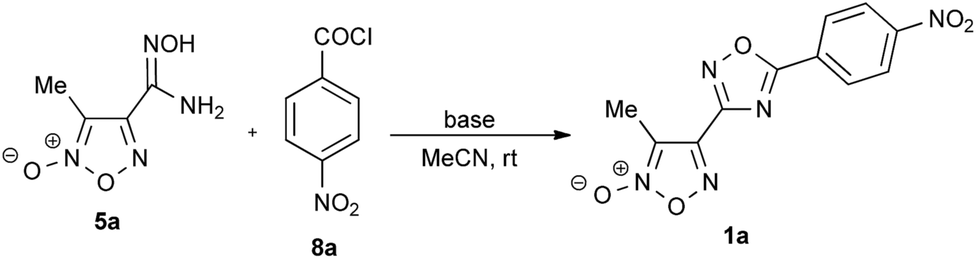
Efficient assembly of mono- and bis(1,2,4-oxadiazol-3-yl)furoxan scaffolds via tandem reactions of furoxanylamidoximes†
Leonid L. Fershtata,
Ivan V. Ananyevb and
Nina N. Makhova*a
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky prosp., 119991 Moscow, Russian Federation. E-mail: mnn@ioc.ac.ru; Fax: +7 499 135 53 28
bA. N. Nesmeyanov Institute of Organoelement Chemistry, Russian Academy of Sciences, 28 Vavilova str., 119991 Moscow, Russian Federation. Fax: +7 499 135 50 85
First published on 20th May 2015
Abstract
A general, facile, highly effective one-pot protocol for the synthesis of new types of heterocyclic systems incorporating mono- and bis(1,2,4-oxadiazol-3-yl)furoxan cores based on the tandem heterocyclization of furoxanylamidoximes with various aliphatic, aromatic, and heterocyclic carboxylic acid chlorides under very mild conditions (Cs2CO3, MeCN, 20 °C) has been developed. In addition, a solvent-free approach for the (1,2,4-oxadiazol-3-yl)furoxan synthesis by the reaction of furoxanylamidoximes with trimethyl orthoformate catalyzed by Sc(OTf)3 has been achieved. The advantages of step economy and scope make these reactions a powerful tool for assembling heterocyclic scaffolds of general chemistry and biomedical interest.
Introduction
A global trend in modern organic chemistry is the design of molecular systems with various degrees of complexity to maximize the incorporation of useful properties while optimizing cost and efficiency.1 To solve this problem, it is necessary to create new, highly effective, room-temperature, regioselective one-pot methods for the synthesis of such structures. One-pot reactions involving two or more reactants lead to expedient assembly of molecules of high structural complexity in a convergent and synthetically efficient manner. These processes avoid the isolation and purification of intermediates, maximize the yield of the final product, minimize the waste of solvent and chromatographic stationary phases, and enhance the greenness of the transformations.2 In particular, one-pot reactions have become an attractive tool in the design of new drug candidates with improved pharmacokinetic profiles by molecular hybridization of different compounds with known pharmacological activity.3Recently,4 we have developed general, facile, room-temperature method for the preparation of previously unknown heterocyclic systems incorporating 1,2,5-oxadiazole 2-oxide (furoxan) ring and various heterocyclic pharmacophores linked by S- and O-bridges. The furoxan moiety has in recent years been the subject of increased attention, pioneered by Gasco owing to a plethora of interesting biological activities related to the ability of furoxans to release NO.5 In particular, a series of hybrid structures representing a combination of various pharmacologically active compounds with furoxan ring, potential NO donor, were synthesized.6 Furthermore, furoxans are of interest as components of energetic formulations due to a positive formation enthalpy and the presence of two oxygen atoms in the ring.7
Among other five-membered heterocycles, 1,2,4-oxadiazole derivatives occupy a special place due to the growing importance of this heterocycle for the design of other heterocycles using their peculiar tendency to undergo molecular rearrangements8 as well as for material construction (polymers, liquid crystals, luminescent materials).9 However, most numerous applications of 1,2,4-oxadiazoles are in the design of biologically active compounds as bioisosters for amide or ester groups10 (anti-diabetic,11 antinflammatory,12 anti-microbial,13 anti-tumoral,14 and neuroprotective15 agents). These biologically active compounds are, as a rule, hybrid molecules containing, apart from the 1,2,4-oxadiazole ring, some aromatic or heterocyclic moieties linked by various aliphatic or heteroatomic spacers. Recently, 1,2,4-oxadiazoles have received attention as components of energetic structures.16
However, hybrid structures incorporating furoxan and 1,2,4-oxadiazole rings are virtually unknown. Only one work was published where 4-amino-3-(1,2,4-oxadiazol-3-yl)furoxan was obtained along with furoxanopyrimidine as a by-product upon treatment of 4-amino-3-(aminohydroxymoyl)furoxan with triethyl orthoformate catalyzed by BF3·Et2O.17
There are two most common routes among the known synthetic strategies for the preparation of the 3,5-disubstituted 1,2,4-oxadiazoles 1 and 1′: (1) [3 + 2]-cycloaddition of nitriles 2 to nitrile oxides 3 and (2) heterocyclization of O-acylamidoximes 4. The latter compounds can be easily prepared by the reaction of nitriles 2 with hydroxylamine followed by reaction of the amidoxime 5 thus formed with activated derivatives of carboxylic acids (chlorides, esters, amides). 3-Monosubstituted 1,2,4-oxadiazoles 6 are prepared by the reaction of amidoximes 5 with trialkyl orthoformates (Scheme 1).
 | ||
| Scheme 1 Various routes to 3-mono- and 3,5-disubstituted 1,2,4-oxadiazoles. | ||
Pathway (1) is seldom applied as nitrile oxides are unstable and are usually prepared in situ in the presence of the corresponding nitriles. Recently,18a we used this approach for the synthesis of 3-nitro-1,2,4-oxadiazoles via [3 + 2]-cycloaddition of activated nitriles to nitroformonitrile oxide generated in situ by the cycloreversion of dinitrofuroxan catalyzed by ionic liquids. The second approach is much more attractive as the nitriles of aliphatic, aromatic, and heterocyclic carboxylic acids are readily accessible.
Here we present the results of our research on the development of general, effective, room-temperature one-pot methods for the preparation of wide series of new hybrid structures: mono- and bis(1,2,4-oxadiazol-3-yl)furoxans by the reaction of furoxanylamidoximes with various carboxylic acid chlorides in the presence of Cs2CO3 as well as with trimethyl orthoformate catalyzed by Sc(OTf)3. Our research group has a great experience in the chemistry of furoxans including the synthesis of effective NO-donors.18
Results and discussion
As the initial compounds, we selected two readily accessible cyanofuroxans: 3-methyl-4-cyanofuroxan 2a19 and 3,4-dicyanofuroxan 2b.20 The initial amidoximes 5a,b were prepared in high yields by the reaction of nitriles 2a,b with hydroxylamine hydrochloride in the presence of K2CO3. The reaction of hydroxylamine with dinitrile 2b involved both nitrile groups (Scheme 2). | ||
| Scheme 2 Synthesis of furoxanylamidoximes 5a,b. | ||
We began our investigations from the search for optimal conditions for the preparation of (1,2,4-oxadiazol-3-yl)furoxans 6a,b by the reaction of amidoximes 5a,b with trimethyl orthoformate. The formation of 1,2,4-oxadiazole ring by similar reactions is usually performed with Lewis acids as catalysts.21 To optimize the reaction conditions, solvent-free reactions of amidoxime 5a with trimethyl orthoformate were carried out in the presence of excess reagent (3.2 mol HC(OMe)3 per mol of 5a) using various Lewis acids catalysts and various temperatures (Table 1). The reaction of substrate 5a with an excess of trimethyl orthoformate in the absence of any catalyst did not result in the formation of desired product 6a (Table 1, entry 1). When 10 mol% of BF3·OEt2 was used as the catalyst at room temperature, no reaction occurred (Table 1, entry 2). The desired product 6a was formed only at 80 °C in moderate yield (Table 1, entry 3). Compound 6a was also obtained with the use of Cu(OTf)2 or PF2(C2F5)3, but the yields were still quite low (38% and 19%, respectively, Table 1, entries 4 and 5). The best catalyst was found to be Sc(OTf)3 (10 mol%). The reaction was completed in 1 minute at 20 °C and resulted in 3-methyl-4-(1,2,4-oxadiazol-3-yl)furoxan 6a formed in 86% yield (Table 1, entry 7).

The designed solvent-free protocol was found to be useful for the preparation of 3,4-bis(1,2,4-oxadiazol-3-yl)furoxan 6b. The Sc(OTf)3-catalyzed reaction of bisamidoxime 5b with trimethyl orthoformate proceeded fast to give the target product in good yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of 3,4-bis(1,2,4-oxadiazol-3-yl)furoxan 6b. | ||
The main efforts of our research were focused on the development of one-pot method for the preparation of previously unknown hybrid molecules containing mono- and bis(1,2,4-oxadiazol-3-yl)furoxan scaffolds with various substituents at C(5) position of the 1,2,4-oxadiazole ring, namely, compounds 1 and 7. For this aim, we investigated cyclocondensation of furoxanylamidoximes 5a,b under the action of aliphatic, aromatic and heteroaromatic carboxylic acid chlorides 8a–k under Lewis acids catalysis or in the presence of bases. The acid chlorides 8 were selected as the most available activated derivatives of carboxylic acids. Two possibilities for the synthesis of 3,5-disubstituted 1,2,4-oxadiazoles from amidoximes and carboxylic acid chlorides are usually applied. The first approach is regarded as a tandem reaction: a mixture of amidoxime 5 and carboxylic acid chloride is treated with a base followed by in situ heating of the formed O-acylamidoxime 4 to induce cyclization into 1,2,4-oxadiazole, e.g., by heating in a sealed tube at 175 °C in pyridine,22 at 130 °C without a solvent,13d at 120 °C in DMF23 or under Lewis acid catalysis.24 For the cyclization of intermediate 4, various bases are added, e.g., DBU,25 TEA,26 or Py.27
The second approach for the construction of 3,5-disubstituted 1,2,4-oxadiazole core involves preliminary isolation of the O-acylamidoximes 4 with the subsequent individual cyclization step proceeding in the presence of a condensation reagent. However, almost in all cases, long-term heating is required for the cyclization to proceed, e.g., refluxing in AcOH,28 in acetone in the presence of K2CO3,29 in dioxane in the presence of KF,30 or in xylene.31 Especially good results were obtained with TBAF as the condensation reagent. In this case, the cyclization is carried out without heating and the final products are formed in rather high yields.32 One example of the 1,2,4-oxadiazole formation upon cyclization of O-acylamidoximes was accomplished at room temperature in the presence of aqueous NaOH.8h Unfortunately, the last-mentioned cyclization procedure is inapplicable in our case due to high sensitivity of furoxan ring to nucleophilic inorganic bases (e.g., alkali).33
To synthesize the desired (1,2,4-oxadiazol-3-yl)furoxans by the reaction of furoxanylamidoximes with carboxylic acid chlorides, we first chose the stepwise protocol. The furoxanylamidoxime 5a and 4-nitrobenzoyl chloride 8a were selected as model compounds. Their interaction in MeCN afforded O-(4-nitrobenzoyl)amidoxime 4a in a nearly quantitative yield even in the absence of any base. Initially, several Lewis acids were investigated in the cyclization step (Table 2, entries 1–5), but the desired product 1a was formed in low yield only with a stoichiometric amount of PF2(C2F5)3 and long-term heating (Table 2, entry 5). A good result was obtained under the action of TBAF, but the yield of the final product was only 54% (Table 2, entry 6). Relying on these results and in view of the known instability of furoxans to strong nucleophiles, we proposed that the use of relatively strong non-nucleophilic bases might be appropriate for the formation of 1,2,4-oxadiazole. Indeed, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) successfully promoted the reaction (Table 2, entry 7). Na2CO3 and K2CO3 in aprotic solvent (MeCN) were effective only at 50 °C (Table 2, entries 8–11), while the replacement of MeCN by DMF resulted in decomposition of initial compound 4a (Table 2, entry 12) due to the increased basicity of the reaction medium. Stronger bases such as Rb2CO3 and Cs2CO3 were efficient at 20 °C (Table 2, entries 13 and 16); however, at higher temperature or with a lower amount of the base, the yields decreased (Table 2, entries 14, 15 and 17). Hence, Cs2CO3 (1.0 equiv.) in MeCN at 20 °C for 1 h was the best for the formation of (1,2,4-oxadiazol-3-yl)furoxan 1a in an excellent yield within a short time and with cost efficiency (Table 2, entry 16).
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | T, °C | Time, h | Yielda (%) |
| a Isolated yields.b 10 mol%.c No reaction.d 20 mol%.e Determined by 1H NMR spectroscopy.f 100 mol%.g Decomposition of compound 4a was observed.h 50 mol%. | |||||
| 1 | Cu(OTf)2b | MeCN | 20 | 72 | —c |
| 2 | Sc(OTf)3b | MeCN | 20 | 72 | —c |
| 3 | PF2(C2F5)3d | MeCN | 80 | 96 | —c |
| 4 | PF2(C2F5)3d | [emim]PF3(C2F5)3 | 100 | 72 | Tracee |
| 5 | PF2(C2F5)3f | [emim]PF3(C2F5)3 | 100 | 72 | 15 |
| 6 | TBAF·3H2Of | Dioxane | 20 | 36 | 54 |
| 7 | DBUf | MeCN | 20 | 1 | 74 |
| 8 | Na2CO3f | MeCN | 20 | 120 | —c |
| 9 | Na2CO3f | MeCN | 50 | 2 | 69 |
| 10 | K2CO3f | MeCN | 20 | 120 | Tracee |
| 11 | K2CO3f | MeCN | 50 | 1 | 76 |
| 12 | K2CO3f | DMF | 20 | 72 | —g |
| 13 | Rb2CO3f | MeCN | 20 | 5 | 87 |
| 14 | Rb2CO3f | MeCN | 50 | 0.25 | 74 |
| 15 | Cs2CO3h | MeCN | 20 | 6 | 42 |
| 16 | Cs2CO3f | MeCN | 20 | 1 | 96 |
| 17 | Cs2CO3f | MeCN | 50 | 0.25 | 53 |

This unanticipated and highly efficient approach to the (1,2,4-oxadiazol-3-yl)furoxan scaffold assembly encouraged us to examine the one-pot synthesis of compound 1a from amidoxime 5a and 4-nitrobenzoyl chloride 8a in the presence of bases that were sufficiently effective for the cyclization of O-acylamidoxime 4a. The reaction was carried out at various molar ratios of the reactants and at various temperatures. The desired product 1a was obtained almost in all cases (Table 3, entries 2–5 and 7–9). However, the best results were again obtained at the presence of Cs2CO3, but for one-pot synthesis of compound 1a, two equivalents of Cs2CO3 were necessary, since one equivalent of Cs2CO3 was spent for neutralization of HCl formed and the second equivalent promoted the cyclization step, and the reaction was completed in 10 hours (TLC monitoring of the disappearance of intermediate 4a) (Table 3, entry 7). An increase or decrease in the amount of Cs2CO3 or temperature rise resulted in a decrease in the yield (Table 3, entries 6, 8 and 9). It is worth mentioning that the reaction times for the one-pot process were longer than the overall time of the two steps in the stepwise protocol. This may be due to the fact that under tandem one-pot reaction conditions, the Cs2CO3 solubility decreased because of the in situ formation of CsCl.
With the optimized conditions in hand, we estimated the range of substrates for the tandem process, and the results are summarized in Table 4. It was established that reaction times depend on the electronic effects of substituents in the initial carboxylic acid chloride. The reactions with aromatic carboxylic acid chlorides possessing electron-withdrawing groups 8a–c and 3-methylfuroxan-4-carboxylic acid chloride 8k (Table 4, entries 1–3, 11) were completed in 10 hours (TLC-monitoring of disappearance of the O-acylamidoxime intermediates, since the Rf values of O-acylamidoximes and 1,2,4-oxadiazoles differ significantly, see the Experimental section for details). Meanwhile, electron-donating groups in the aromatic ring and aliphatic substituents in acyl chlorides slowed down the reaction, but the yields of the final products were still good (Table 4, entries 5–7). To our delight, heterocyclic carboxylic acid chlorides containing furan, pyridine or pyrazole moieties reacted smoothly under the optimized conditions to give corresponding tandem products 1h–j in high yields (Table 4, entries 8–10).






















Next, we extended our approach to bis(amidoxime) 5b. First, we have studied the stepwise protocol for the synthesis of 3,4-bis[5-(4-nitrophenyl)-1,2,4-oxadiazol-3-yl]furoxan 7a. The acylation step proceeded fast with the formation of 3,4-bis(O-4-nitrobenzoyl)amidoxime 4b in a nearly quantitative yield. The cyclization step was performed with two equivalents of Cs2CO3 and resulted in the formation of bis(oxadiazolyl)furoxan 7a in excellent yield (Scheme 4).
 | ||
| Scheme 4 Cyclization of 3,4-bis(O-4-nitrobenzoyl)amidoxime 4b. | ||
Being encouraged by this result, we proceeded to one-pot synthesis of compounds 7a–k from bis(amidoxime) 5b and carboxylic acid chlorides 8a–k (Table 5). It was found that duration of the reaction between amidoxime 5b and carboxylic acid chloride 8a was significantly greater (24 hours) than the duration of the similar reaction of amidoxime 5a (10 hours, see Table 4) (TLC monitoring of disappearance of the O-acylamidoxime intermediates, since the Rf values of O-acylamidoximes and 1,2,4-oxadiazoles differ significantly, see Experimental section for details). Evidently, this difference is caused by a special feature of the furoxan ring structure. It is known that the C(3) carbon atom of the furoxan ring has a higher electron density than C(4) due to the resonance influence of the N-oxide oxygen atom.34 An increase in the electron density on C(3) can result in higher electron density on the NH2 group of intermediate 4b and, hence, slow down the cyclization. Nevertheless, we succeeded in realization of the tandem one-pot protocol for the synthesis of 3,4-bis(1,2,4-oxadiazol-3-yl)furoxans 7a–k containing aliphatic, aromatic, and heterocyclic substituents at the C(5) position of the 1,2,4-oxadiazole ring in good to excellent yields (70–89%, Table 5).












A plausible mechanism for the Cs2CO3-mediated reaction of furoxanylamidoximes 5 with carboxylic acid chlorides 8 is outlined in Scheme 5. It seems that Cs2CO3 acts both as a base and a dehydrating reagent. Since Cs2CO3 is a strong base, one could expect the amino group deprotonation in O-acylamidoxime 4 with subsequent intramolecular nucleophilic cyclization in intermediate 9 to give the cesium salt of dihydro-1,2,4-oxadiazol-5-ol 10. Cationic metathesis of salt 10 with CsHCO3 results in “free” dihydro-1,2,4-oxadiazol-5-ol 11 and regeneration of Cs2CO3, which mediates the final dehydration step.
 | ||
| Scheme 5 Plausible mechanism for the formation of (1,2,4-oxadiazol-3-yl)furoxans. | ||
All of the synthesized (1,2,4-oxadiazol-3-yl)furoxans were characterized by spectral and analytical methods. Finally, we confirmed the structures of the 1,2,4-oxadiazole derivatives by a single-crystal X-ray diffraction study of the representative compound 1d (Fig. 1).
 | ||
| Fig. 1 The general view of the 1d molecule. Atoms are represented by probability ellipsoids of atomic vibrations (ρ = 50%). | ||
The 1d molecule are nearly flat in crystal with the only exceptions of hydrogen atoms of the methyl group: the C(10)C(5)C(4)O(3) and N(3)C(3)C(2)C(1) torsion angles are 5.1(3) and 2.5(3)° correspondingly, while the maximum deviation from the mean-square plane composed by all non-hydrogen atoms is 0.11(2) Å (for the C(9) atom). Being in line with rather short C(2)–C(3) and C(4)–C(5) bonds (1.465(3) and 1.461(3) Å, respectively) demonstrates a significant contribution of π-conjugation into the stabilization of molecule structure. The bond lengths distribution within heterocyclic fragments can be considered as expected for this class of compounds. In crystal molecules are stuck together by shortened O⋯π contacts between furoxan cycles (O(1)⋯C(1) 2.956(3) Å) and C–H⋯π contacts between the benzene ring and the furoxan fragment (C(8)⋯O(1) 3.310(3), with C–H bond length being normalized on 1.080 Å H(8)⋯O(1) 2.477 Å, angle C(8)–H(8)⋯O(1) – 133°) and form layers. The three-dimensional crystal structure is formed by weak H⋯H interactions between benzene rings (Fig. 2).
 | ||
| Fig. 2 A fragment of the crystal packing of 1d, demonstrating layer-type structure. Non-covalent intermolecular O⋯π and C–H⋯π interactions are shown by dash lines. | ||
Conclusion
In summary, facile, effective, general, room-temperature one-pot methods for the construction of previously unknown heterocyclic systems containing 4-mono- and 3,4-bis(5-R-1,2,4-oxadiazol-3-yl)furoxans by tandem heterocyclization of the furoxanylamidoximes with aliphatic, aromatic, and heteroaromatic carboxylic acid chlorides under very mild conditions have been developed. The target compounds were prepared in good to excellent yields. To the best of our knowledge, our study is the first example of the application of Cs2CO3 in the synthesis of 1,2,4-oxadiazole derivatives. In addition, the possibility to obtain 4-mono- and 3,4-bis(1,2,4-oxadiazol-3-yl)furoxans in high yields has been exemplified by the solvent-free reaction of the furoxanylamidoximes with trimethyl orthoformate catalyzed by Sc(OTf)3. The advantages of these new methods are operational simplicity, step economy, and the use of environmentally friendly Cs2CO3 and Sc(OTf)3. The developed methods provide a powerful tool for the synthesis of an extensive series of new types of hybrid molecules – (1,2,4-oxadiazol-3-yl)furoxans, including heterocyclic sequences containing five heterocyclic fragments, e.g. a structure consisting of three furoxan rings linked by two 1,2,4-oxadiazole bridges. The study of cytotoxic activity of the synthesized compounds is now in progress.Experimental section
General remarks
All reactions were carried out in well-cleaned oven-dried glassware with magnetic stirring. 1H and 13C NMR spectra were recorded on a Bruker AM-300 (300.13 and 75.47 MHz, respectively) and Bruker AC-200 (200.13 and 50.32 MHz, respectively) and referenced to residual solvent peak. The chemical shifts are reported in ppm (δ); multiplicities are indicated by s (singlet), d (doublet), t (triplet), m (multiplet) and br (broad). Coupling constants, J, are reported in Hertz. The IR spectra were recorded on a Bruker “Alpha” spectrometer in the range 400–4000 cm−1 (resolution 2 cm−1) as pellets with KBr or as a thin layer. Elemental analyses were performed by the CHN Analyzer Perkin-Elmer 2400. The melting points were determined on Kofler melting point apparatus and are uncorrected. Analytical thin-layer chromatography (TLC) was carried out on Merck 25 TLC silica gel 60 F254 aluminum sheets. The visualization of the TLC plates was accomplished with a UV light.High resolution mass spectra were recorded on a Bruker microTOF spectrometer with electrospray ionization (ESI). All measurements were performed in a positive (+MS) ion mode (interface capillary voltage: 4500 V) with scan range m/z: 50–3000. External calibration of the mass spectrometer was performed with Electrospray Calibrant Solution (Fluka). A direct syringe injection was used for all analyzed solutions in MeCN (flow rate: 3 μL min−1). Nitrogen was used as nebulizer gas (0.4 bar) and dry gas (4.0 L min−1); interface temperature was set at 180 °C. All spectra were processed by using Bruker DataAnalysis 4.0 software package. MeCN (HPLC grade) for ESI-HRMS experiments was ordered from Merck and used as supplied. All samples for ESI-HRMS experiments were prepared in 1.5 mL Eppendorf tubes. All plastic disposables (Eppendorf tubes and tips) used in sample preparation were washed with MeCN before use.
3-Methyl-4-cyanofuroxan 2a was prepared according to the method described in ref. 19 and 3,4-dicyanofuroxan 2b was prepared according to the method described in ref. 20. Carboxylic acid chlorides were prepared according to a known procedure.36 MeCN and CH2Cl2 were distilled before use over the corresponding drying agents. All other reagents were purchased from Aldrich and used without further purification.
Crystallographic data
Crystals of 1d (C11H8N4O3, M = 244.21) are monoclinic, space group P21/n, at 120 K: a = 6.5740(13), b = 22.414(5), c = 7.9769(16), β = 113.126(4), V = 1080.9(4) Å3, Z = 4 (Z′ = 1), dcalc = 1.501 g cm−3, μ(MoKα) = 0.90 cm−1, F(000) = 2664. Intensities of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 165 reflections were measured with a Bruker SMART APEX2 CCD diffractometer [λ(MoKα) = 0.71072 Å, ω-scans, 2θ < 56°] and 2625 independent reflections [Rint = 0.0924] were used in further refinement. The structure was solved by direct method and refined by the full-matrix least-squares technique against F2 in the isotropic–anisotropic approximation. The hydrogen atoms positions were calculated; hydrogen atoms were refined in the isotropic approximation within the riding model. For 1d, the refinement converged to wR2 = 0.1429 and GOF = 1.032 for all independent reflections (R1 = 0.0625 was calculated against F for 1671 observed reflections with I > 2σ(I)). All calculations were performed using SHELXTL PLUS 5.0 (ESI†).35
165 reflections were measured with a Bruker SMART APEX2 CCD diffractometer [λ(MoKα) = 0.71072 Å, ω-scans, 2θ < 56°] and 2625 independent reflections [Rint = 0.0924] were used in further refinement. The structure was solved by direct method and refined by the full-matrix least-squares technique against F2 in the isotropic–anisotropic approximation. The hydrogen atoms positions were calculated; hydrogen atoms were refined in the isotropic approximation within the riding model. For 1d, the refinement converged to wR2 = 0.1429 and GOF = 1.032 for all independent reflections (R1 = 0.0625 was calculated against F for 1671 observed reflections with I > 2σ(I)). All calculations were performed using SHELXTL PLUS 5.0 (ESI†).35
Synthesis of amidoximes 5a,b
A mixture of furoxancarbonitrile 2a,b (6.48 mmol), hydroxylamine hydrochloride (0.68 g, 9.72 mmol for substrate 2a and 1.35 g, 19.44 mmol for substrate 2b) and K2CO3 (1.34 g, 9.72 mmol for substrate 2a and 2.68 g, 19.44 mmol for substrate 2b) in water (15 mL) was stirred for 30–60 min at room temperature. The solid formed was filtered off, washed with water and dried in air to afford amidoximes 5a,b.:EtOAc = 10:1). 1H NMR (200 MHz, DMSO-d6) δH: 2.26 (3H, s, Me), 5.47 (2H, br. s, NH2), 10.50 (1H, br. s, NOH). 13C NMR (75.5 MHz, DMSO-d6) δC: 8.75 (Me), 109.20 (C-3 furoxan), 139.13 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH), 150.81 (C-4 furoxan). IR (KBr): 3479, 3376, 3000, 2891, 1687, 1660, 1610, 1537, 1462, 1361, 1281, 1176, 1124, 1048, 935, 870, 795, 639 cm−1. Calcd for C4H6N4O3 (%): C, 30.38; H, 3.82; N, 35.43. Found (%): C, 30.31; H, 3.86; N, 35.36.:EtOAc = 5:1). 1H NMR (300 MHz, DMSO-d6) δH: 6.09 (4H, br. s, 2NH2), 10.09 (1H, br. s, NOH), 10.65 (1H, br. s, NOH). 13C NMR (75.5 MHz, DMSO-d6) δC: 109.25 (C-3 furoxan), 139.10 (CNOH), 141.80 (CNOH), 150.88 (C-4 furoxan). IR (KBr): 3464, 3373, 3314, 2872, 1650, 1583, 1539, 1503, 1419, 1363, 1313, 1230, 1100, 1022, 954, 750 cm−1. Calcd for C4H6N6O4 (%): C, 23.77; H, 2.99; N, 41.58. Found (%): C, 23.72; H, 3.07; N, 41.64.
NOH), 150.81 (C-4 furoxan). IR (KBr): 3479, 3376, 3000, 2891, 1687, 1660, 1610, 1537, 1462, 1361, 1281, 1176, 1124, 1048, 935, 870, 795, 639 cm−1. Calcd for C4H6N4O3 (%): C, 30.38; H, 3.82; N, 35.43. Found (%): C, 30.31; H, 3.86; N, 35.36.:EtOAc = 5:1). 1H NMR (300 MHz, DMSO-d6) δH: 6.09 (4H, br. s, 2NH2), 10.09 (1H, br. s, NOH), 10.65 (1H, br. s, NOH). 13C NMR (75.5 MHz, DMSO-d6) δC: 109.25 (C-3 furoxan), 139.10 (CNOH), 141.80 (CNOH), 150.88 (C-4 furoxan). IR (KBr): 3464, 3373, 3314, 2872, 1650, 1583, 1539, 1503, 1419, 1363, 1313, 1230, 1100, 1022, 954, 750 cm−1. Calcd for C4H6N6O4 (%): C, 23.77; H, 2.99; N, 41.58. Found (%): C, 23.72; H, 3.07; N, 41.64.Synthesis of (1,2,4-oxadiazol-3-yl)furoxans 6a,b
Sc(OTf)3 (0.049 g, 0.1 mmol) was added at room temperature to the stirred suspension of amidoxime 5a,b (1 mmol) in (0.3 mL for substrate 5a and 0.6 mL for substrate 5b). After 1–5 min the reaction mixture became homogenous and H2O (6 mL) was added. The solid formed was filtered off, washed with water and dried in air to afford 1,2,4-oxadiazoles 6a,b.:EtOAc = 10:1). 1H NMR (200 MHz, CDCl3) δH: 2.49 (3H, s, Me), 9.04 (1H, s, CH). 13C NMR (50 MHz, CDCl3) δC: 8.66, 111.38, 146.50, 159.28, 165.92. IR (KBr): 2923, 1624, 1583, 1546, 1468, 1341, 1272, 1123, 856, 748, 716 cm−1. Calcd for C5H4N4O3 (%): C, 35.72; H, 2.40; N, 33.33. Found (%): C, 35.63; H, 2.47; N, 33.39.:EtOAc = 5:1). 1H NMR (300 MHz, DMSO-d6) δH: 9.90 (1H, s, CH), 9.96 (1H, s, CH). 13C NMR (50 MHz, DMSO-d6) δC: 106.14, 145.17, 155.44, 157.50, 168.19, 168.39. IR (KBr): 2931, 1628, 1581, 1542, 1433, 1352, 1276, 1116, 851, 741 cm−1. Calcd for C6H2N6O4 (%): C, 32.44; H, 0.91; N, 37.84. Found (%): C, 32.37; H, 0.96; N, 37.91.General procedure for the synthesis of O-(4-nitrobenzoyl)amidoximes 4a,b
To the solution of amidoxime 5a,b (1.0 mmol) in MeCN (3 mL) at stirring and room temperature 4-nitrobenzoyl chloride 8a (1.0 mmol for substrate 5a and 2.0 mmol for substrate 5b) was added. The mixture was stirred for 3–5 min (TLC monitoring, eluent CHCl3:EtOAc = 10:1 or 5:1), the solid formed was filtered off, washed with water, then with MeCN and dried in air.
:EtOAc = 10:1). 1H NMR (200 MHz, DMSO-d6) δH: 2.36 (3H, s, Me), 7.61 (2H, s, NH2), 8.32 (2H, d, 3J = 8.6 Hz, H Ar), 8.48 (2H, d, 3J = 8.6 Hz, H Ar). 13C NMR (75.5 MHz, DMSO-d6) δC: 8.65, 112.45, 124.57, 127.69, 129.72, 147.38, 150.27, 160.28, 174.64. Calcd for C11H9N5O6 (%): C, 43.00; H, 2.95; N, 22.80. Found (%): C, 42.92; H, 3.02; N, 22.86.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 7.61 (4H, s, 2NH2), 8.25 (4H, d, 3J = 6.4 Hz, H Ar), 8.40 (4H, d, 3J = 6.4 Hz, H Ar). 13C NMR (75.5 MHz, DMSO-d6) δC: 108.23, 123.09, 130.93, 130.95, 131.00, 131.02, 133.90, 145.72, 147.43, 149.27, 150.06, 150.08, 161.23, 161.35. Calcd for C18H12N8O10 (%): C, 43.21; H, 2.42; N, 22.40. Found (%): C, 43.15; H, 2.49; N, 22.33.General procedure for the synthesis of 1,2,4-oxadiazoles 1a–k
The mixture of amidoxime 5a (1.0 mmol), carboxylic acid chloride (1.0 mmol) and Cs2CO3 (2.0 mmol, 652 mg) in MeCN (3 mL) was stirred for 10–24 h at room temperature. Then H2O (10 mL) was added. The solid formed was filtered off, washed with water and dried in air to afford 1,2,4-oxadiazoles 1a–e,h–k. Compounds 1f,g were extracted with CH2Cl2 (3 × 5 mL), the combined organic phase was washed with H2O (15 mL) and dried over MgSO4.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.44 (3H, s, Me), 8.46 (4H, s, H Ar). 13C NMR (50 MHz, DMSO-d6) δC: 8.79, 112.59, 124.70, 127.78, 129.83, 147.50, 150.36, 160.40, 174.74. IR (KBr): 1619, 1564, 1530, 1472, 1345, 1279, 1129, 855, 747, 711, 639 cm−1. Calcd for C11H7N5O5 (%): C, 45.68; H, 2.44; N, 24.22. Found (%): C, 45.61; H, 2.48; N, 24.27.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.46 (3H, s, Me), 7.98–8.03 (1H, m, H Ar), 8.59–8.64 (2H, m, H Ar), 8.86 (1H, s, H Ar). 13C NMR (75.5 MHz, DMSO-d6) δC: 8.76, 112.59, 122.73, 123.95, 128.16, 131.63, 134.19, 147.51, 148.29, 160.28, 174.66. IR (KBr): 1623, 1597, 1562, 1527, 1345, 1130, 1037, 929, 847, 747, 713, 635 cm−1. HRMS (ESI) m/z for C11H7N5NaO5 (M + Na)+: calcd 312.0339, found 312.0346. Calcd for C11H7N5O5 (%): C, 45.68; H, 2.44; N, 24.22. Found (%): C, 45.60; H, 2.51; N, 24.29.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.41 (3H, s, Me), 8.02–8.05 (2H, m, H Ar), 8.20–8.23 (1H, m, H Ar), 8.27–8.30 (1H, m, H Ar). 13C NMR (50 MHz, DMSO-d6) δC: 8.69, 112.61, 116.47, 125.08, 131.85, 134.02, 134.81, 147.32, 148.06, 160.08, 173.54. IR (KBr): 1631, 1617, 1564, 1525, 1492, 1423, 1354, 1095, 1043, 925, 848, 787, 749, 720, 634 cm−1. Calcd for C11H7N5O5 (%): C, 45.68; H, 2.44; N, 24.22. Found (%): C, 45.62; H, 2.37; N, 24.16.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.43 (3H, s, Me), 7.69–7.76 (3H, m, H-3,4,5 Ph), 8.21 (2H, br. s, H-2,6 Ph). 13C NMR (50 MHz, DMSO-d6) δC: 8.53, 112.39, 122.20, 127.93, 129.45, 133.78, 147.45, 159.87, 176.01. IR (KBr): 1624, 1607, 1557, 1494, 1426, 1350, 1124, 1037, 952, 923, 852, 750, 710, 686, 639 cm−1. Calcd for C11H8N4O3 (%): C, 54.10; H, 3.30; N, 22.94. Found (%): C, 54.03; H, 3.36; N, 22.99.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.39 (3H, s, Me), 3.87 (3H, s, OMe), 7.18 (2H, d, 3J = 8.1 Hz, H Ar), 8.11 (2H, d, 3J = 8.1 Hz, H Ar). 13C NMR (75.5 MHz, DMSO-d6) δC: 9.36, 56.24, 113.15, 115.24, 115.62, 130.86, 148.28, 160.54, 164.27, 176.67. IR (KBr): 1608, 1560, 1506, 1429, 1347, 1309, 1257, 1177, 1124, 1109, 1068, 1021, 951, 846, 765, 642 cm−1. Calcd for C12H10N4O4 (%): C, 52.56; H, 3.68; N, 20.43. Found (%): C, 52.51; H, 3.74; N, 20.37.:EtOAc = 10:1). 1H NMR (200 MHz, CDCl3) δH: 2.47 (3H, s, Me), 3.55 (3H, s, OMe), 4.81 (2H, s, OCH2). 13C NMR (50 MHz, CDCl3) δC: 8.62, 59.64, 64.77, 111.31, 146.64, 159.74, 177.42. IR (thin layer with KBr): 2938, 2834, 1619, 1577, 1483, 1431, 1344, 1197, 1117, 1040, 950, 920, 848, 633 cm−1. Calcd for C7H8N4O4 (%): C, 39.63; H, 3.80; N, 26.41. Found (%): C, 39.56; H, 3.85; N, 26.36.:EtOAc = 10:1). Yield 225 mg (78%). 1H NMR (200 MHz, CDCl3) δH: 2.49 (3H, s, Me), 4.75 (2H, s, CH2OCH2Ph), 4.88 (2H, s, CH2OCH2Ph), 7.39 (s, 5H, Ph). 13C NMR (50 MHz, CDCl3) δC: 8.68, 62.04, 73.74, 111.32, 127.99, 128.30, 128.52, 135.97, 146.68, 159.80, 177.51. IR (thin layer with KBr): 3025, 2886, 2861, 1615, 1573, 1485, 1431, 1345, 1267, 1121, 1098, 1037, 950, 914, 851, 751, 699, 638 cm−1. HRMS (ESI) m/z for C13H12N4NaO4 (M + Na)+: calcd 311.0751, found 311.0745. Calcd for C13H12N4O4 (%): C, 54.17; H, 4.20; N, 19.44. Found (%): C, 54.11; H, 4.27; N, 19.49.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.41 (3H, s, Me), 6.92 (1H, br. s, H Het), 7.77 (1H, br. s, H Het), 8.23 (1H, br. s, H Het). 13C NMR (50 MHz, DMSO-d6) δC: 8.79, 112.64, 113.45, 119.16, 138.29, 147.56, 149.28, 159.90, 167.95. IR (KBr): 3149, 3130, 1615, 1543, 1468, 1428, 1344, 1287, 1238, 1164, 1126, 1038, 1025, 978, 899, 851, 778, 635 cm−1. Calcd for C9H6N4O4 (%): C, 46.16; H, 2.58; N, 23.93. Found (%): C, 46.11; H, 2.64; N, 23.98.:EtOAc = 10:1). 1H NMR (200 MHz, DMSO-d6) δH: 2.44 (3H, s, Me), 8.13 (2H, br. s, H Het), 8.93 (2H, br. s, H Het). 13C NMR (50 MHz, DMSO-d6) δC: 8.80, 112.61, 121.42, 129.53, 147.55, 151.31, 160.39, 174.86. IR (KBr): 3048, 1625, 1573, 1544, 1493, 1417, 1349, 1282, 1125, 1080, 1036, 951, 930, 844, 760, 695, 637 cm−1. Calcd for C10H7N5O3 (%): C, 48.98; H, 2.88; N, 28.56. Found (%): C, 49.04; H, 2.81; N, 28.64.:EtOAc = 10:1). 1H NMR (200 MHz, DMSO-d6) δH: 2.42 (3H, s, Me in furoxan), 2.69 (3H, s, Me in pyrazole), 7.60 (5H, s, Ph), 8.42 (1H, s, CH). 13C NMR (75.5 MHz, DMSO-d6) δC: 9.34, 12.47, 105.92, 113.21, 125.85, 129.59, 130.03, 138.68, 140.72, 143.27, 148.30, 160.21, 172.82. IR (KBr): 3110, 3080, 1617, 1576, 1499, 1455, 1403, 1379, 1342, 1233, 1119, 1034, 935, 845, 769, 699, 634 cm−1. Calcd for C15H12N6O3 (%): C, 55.55; H, 3.73; N, 25.91. Found (%): C, 55.61; H, 3.64; N, 25.97.:EtOAc = 10:1). 1H NMR (300 MHz, DMSO-d6) δH: 2.43 (3H, s, Me), 2.47 (3H, s, Me). 13C NMR (50 MHz, DMSO-d6) δC: 8.58, 8.78, 112.46, 112.67, 145.80, 147.14, 160.14, 166.64. IR (KBr): 1620, 1579, 1499, 1491, 1434, 1385, 1342, 1105, 1043, 995, 951, 926, 850, 766, 670, 632 cm−1. HRMS (ESI) m/z for C8H6N6NaO5 (M + Na)+: calcd 289.0292, found 289.0299. Calcd for C8H6N6O5 (%): C, 36.10; H, 2.27; N, 31.57. Found (%): C, 36.02; H, 2.35; N, 31.64.General procedure for the synthesis of 1,2,4-oxadiazoles 7a–k
The mixture of amidoxime 5b (1.0 mmol), carboxylic acid chloride (2.0 mmol) and Cs2CO3 (4.0 mmol, 1.304 g) in MeCN (5 mL) was stirred for 24–48 h at room temperature. Then H2O (10 mL) was added. The solid formed was filtered off, washed with water and dried in air to afford 1,2,4-oxadiazoles 7a–e,h–k. Compounds 7f,g were extracted with CH2Cl2 (3 × 5 mL), the combined organic phase was washed with H2O (15 mL) and dried over MgSO4.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 8.36–8.47 (8H, m, H Ar). 13C NMR (50 MHz, DMSO-d6) δC: 112.14, 124.66, 125.03, 127.52, 127.73, 129.79, 130.63, 148.16, 150.32, 150.86, 160.36, 160.68, 172.86, 174.69. IR (KBr): 3109, 3083, 1615, 1570, 1525, 1347, 1269, 869, 854, 734, 719 cm−1. Calcd for C18H8N8O8 (%): C, 46.56; H, 1.74; N, 24.13. Found (%): C, 46.49; H, 1.67; N, 24.07.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 7.88 (2H, br. s, H Ar), 8.49 (4H, br. s, H Ar), 8.78 (1H, br. s, H Ar), 8.88 (1H, br. s, H Ar). 13C NMR (50 MHz, DMSO-d6) δC: 105.36, 122.70, 123.88, 123.96, 126.49, 127.84, 129.66, 131.18, 133.67, 133.75, 134.35, 144.90, 148.14, 148.21, 156.98, 159.09, 174.59, 174.79. IR (KBr): 3087, 1633, 1532, 1391, 1349, 1291, 1261, 1127, 1075, 1030, 947, 912, 872, 817, 740, 717, 667 cm−1. HRMS (ESI) m/z for C18H8N8NaO8 (M + Na)+: calcd 487.0357, found 487.0337. Calcd for C18H8N8O8 (%): C, 46.56; H, 1.74; N, 24.13. Found (%): C, 46.62; H, 1.66; N, 24.06.:EtOAc = 5:1). 1H NMR (300 MHz, DMSO-d6) δH: 7.61–7.87 (2H, m, H Ar), 8.02–8.29 (6H, m, H Ar). 13C NMR (50 MHz, DMSO-d6) δC: 105.60, 115.81, 116.15, 124.59, 125.35, 131.47, 131.85, 132.04, 133.84, 134.02, 134.81, 135.64, 148.04, 149.06, 149.35, 156.78, 162.09, 173.35. IR (KBr): 2924, 1642, 1618, 1537, 1481, 1431, 1362, 1099, 1012, 913, 853, 793, 751, 636 cm−1. Calcd for C18H8N8O8 (%): C, 46.56; H, 1.74; N, 24.13. Found (%): C, 46.51; H, 1.69; N, 24.17.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 7.66–7.70 (6H, m, H-3,4,5 Ph), 8.11–8.16 (4H, m, H-2,6 Ph). 13C NMR (75.5 MHz, DMSO-d6) δC: 106.17, 122.31, 122.42, 128.15, 128.32, 129.55, 129.81, 132.32, 134.05, 145.55, 156.92, 158.98, 176.53. IR (KBr): 3066, 1624, 1607, 1559, 1490, 1450, 1392, 1270, 1236, 1051, 1029, 976, 947, 811, 752, 714, 687 cm−1. Calcd for C18H10N6O4 (%): C, 57.76; H, 2.69; N, 22.45. Found (%): C, 57.83; H, 2.74; N, 22.37.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 3.85 (6H, s, 2 OCH3), 7.04 (4H, d, 3J = 8.0 Hz, H Ar), 8.15 (4H, d, 3J = 8.0 Hz, H Ar). 13C NMR (50 MHz, DMSO-d6) δC: 55.50, 56.30, 113.89, 115.18, 115.73, 120.62, 130.21, 130.32, 131.86, 132.14, 133.39, 144.74, 146.84, 151.34, 162.49, 163.27. IR (KBr): 1612, 1572, 1513, 1444, 1331, 1282, 1221, 1184, 1101, 1068, 1024, 853, 772, 657 cm−1. Calcd for C20H14N6O6 (%): C, 55.30; H, 3.25; N, 19.35. Found (%): C, 55.36; H, 3.34; N, 19.27.:EtOAc = 5:1). 1H NMR (300 MHz, DMSO-d6) δH: 3.42 (3H, s, OMe), 3.45 (3H, s, OMe), 4.83 (2H, s, OCH2), 4.88 (2H, s, OCH2). 13C NMR (50 MHz, DMSO-d6) δC: 58.85, 64.28, 64.33, 105.81, 145.27, 156.10, 158.18, 177.78, 178.04. IR (thin layer with KBr): 2939, 2833, 1632, 1578, 1451, 1366, 1274, 1197, 1121, 970, 919, 816 cm−1. Calcd for C10H10N6O6 (%): C, 38.72; H, 3.25; N, 27.09. Found (%): C, 38.79; H, 3.32; N, 27.01.:EtOAc = 5:1). 1H NMR (300 MHz, DMSO-d6) δH: 4.67 (2H, s, CH2OCH2Ph), 4.70 (2H, s, CH2OCH2Ph), 4.79 (2H, s, CH2OCH2Ph), 4.82 (2H, s, CH2OCH2Ph), 7.35 (10H, s, 2 Ph). 13C NMR (50 MHz, DMSO-d6) δC: 61.96, 73.57, 104.95, 128.03, 128.30, 128.52, 135.93, 144.75, 156.32, 158.51, 177.64. Calcd for C22H18N6O6 (%): C, 57.14; H, 3.92; N, 18.17. Found (%): C, 57.22; H, 3.85; N, 18.09.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 6.88–6.92 (2H, m, H Het), 7.67–7.79 (2H, m, H Het), 8.20–8.22 (2H, m, H Het). 13C NMR (50 MHz, DMSO-d6) δC: 112.05, 112.26, 113.45, 119.21, 119.27, 138.07, 138.18, 148.19, 149.22, 149.34, 156.73, 158.71, 167.97, 168.19. IR (KBr): 3127, 1622, 1539, 1523, 1469, 1406, 1378, 1286, 1235, 1216, 1175, 1106, 1073, 1017, 945, 901, 761, 591 cm−1. Calcd for C14H6N6O6 (%): C, 47.47; H, 1.71; N, 23.72. Found (%): C, 47.53; H, 1.62; N, 23.81.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 8.13 (4H, br. s, H Het), 8.95 (4H, br. s, H Het). 13C NMR (50 MHz, DMSO-d6) δC: 111.13, 120.61, 121.38, 129.48, 130.19, 148.67, 151.27, 151.46, 159.84, 160.39, 172.13, 174.86. Calcd for C16H8N8O4 (%): C, 51.07; H, 2.14; N, 29.78. Found (%): C, 50.99; H, 2.22; N, 29.69.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 2.70 (6H, s, CH3), 7.59–7.67 (10H, m, Ph), 8.48 (2H, s, 2 CH). 13C NMR (50 MHz, DMSO-d6) δC: 12.47, 12.72, 105.76, 105.92, 112.68, 113.21, 125.64, 125.85, 129.12, 129.59, 130.02, 130.64, 138.32, 138.68, 140.16, 140.72, 143.27, 143.75, 148.62, 160.01, 160.20, 172.81, 173.36. Calcd for C26H18N10O4 (%): C, 58.43; H, 3.39; N, 26.21. Found (%): C, 58.36; H, 3.47; N, 26.30.:EtOAc = 5:1). 1H NMR (200 MHz, DMSO-d6) δH: 2.35 (3H, s, Me), 2.43 (3H, s, Me). 13C NMR (50 MHz, DMSO-d6) δC: 8.32, 8.48, 109.25, 112.37, 112.40, 144.63, 145.61, 145.71, 157.12, 158.94, 166.43, 166.88. IR (KBr): 1624, 1586, 1493, 1481, 1431, 1389, 1321, 1115, 1040, 995, 854, 672, 634 cm−1. Calcd for C12H6N10O8 (%): C, 34.46; H, 1.45; N, 33.49. Found (%): C, 34.39; H, 1.56; N, 33.43.Acknowledgements
L.L. Fershtat and N.N. Makhova are gratefully acknowledged to the Russian Science Foundation for the financial support towards their experimental research (Project no. 14-50-00126). I.V. Ananyev thanks Russian Science Foundation (Project no. 14-13-00884) for the financial support of the X-ray structural studies.Notes and references
- (a) V. P. Ananikov, E. A. Khokhlova, M. P. Egorov, A. M. Sakharov, S. G. Zlotin, A. V. Kucherov, L. M. Kustov, M. L. Gening and N. E. Nifantiev, Mendeleev Commun., 2015, 25, 75 CrossRef CAS PubMed; (b) V. P. Ananikov, L. L. Khemchyan, Yu. V. Ivanova, V. I. Bukhtiyarov, A. M. Sorokin, I. P. Prosvirin, S. Z. Vatsadze, A. V. Medved'ko, V. N. Nuriev, A. D. Dilman, V. V. Levin, I. V. Koptyug, K. V. Kovtunov, V. V. Zhivonitko, V. A. Likholobov, A. V. Romanenko, P. A. Simonov, V. G. Nenajdenko, O. I. Shmatova, V. M. Muzalevskiy, M. S. Nechaev, A. F. Asachenko, O. S. Morozov, P. B. Dzhevakov, S. N. Osipov, D. V. Vorobyeva, M. A. Topchiy, M. A. Zotova, S. A. Ponomarenko, O. V. Borshchev, Yu. N. Luponosov, A. A. Rempel, A. A. Valeeva, A. Yu. Stakheev, O. V. Turova, I. S. Mashkovsky, S. V. Sysolyatin, V. V. Malykhin, G. A. Bukhtiyarova, A. O. Terent'ev and I. B. Krylov, Russ. Chem. Rev., 2014, 83, 885 CrossRef PubMed; (c) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (d) B. M. Trost, Acc. Chem. Res., 2002, 35, 695 CrossRef CAS PubMed; (e) B. M. Trost, Science, 1991, 254, 1471 CAS; (f) A. A. Tabolin, R. A. Novikov, Yu. A. Khomutova, A. A. Zharov, G. A. Stashina, Yu. V. Nelyubina, Yu. V. Tomilov and S. L. Ioffe, Tetrahedron Lett., 2015, 56, 2102 CrossRef CAS PubMed.
- For recent reviews on the molecular complexity and one-pot reactions, see: (a) K. C. Nicolaou, C. R. H. Hale, C. Nilewski and H. A. Ioannidou, Chem. Soc. Rev., 2012, 41, 5185 RSC; (b) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234 CrossRef CAS PubMed; (c) N. Isambert and R. Lavila, Chem.–Eur. J., 2008, 14, 8444 CrossRef CAS PubMed; (d) V. Jeena and R. S. Robinson, RSC Adv., 2014, 4, 40720 RSC; (e) N. Arumugam, R. S. Kumar, A. I. Almansour and S. Perumal, Curr. Org. Chem., 2013, 17, 1929 CrossRef CAS.
- For selected examples on the use of molecular hybridization approach in drug design, see: (a) M. S. Shaikh, M. B. Palkar, H. M. Patel, R. A. Rane, W. S. Alwan, M. M. Shaikh, I. M. Shaikh, G. A. Hampannavar and R. Karpoormath, RSC Adv., 2014, 4, 62308 CAS; (b) P. L. Bosquesi, T. R. F. Melo, E. O. Vizioli, J. L. Dos Santos and M. C. Chung, Pharmaceuticals, 2011, 4, 1450 CrossRef CAS PubMed; (c) R. C. Maia and C. A. M. Fraga, Curr. Enzyme Inhib., 2010, 6, 171 CrossRef CAS; (d) C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E. J. Barreiro and C. A. M. Fraga, Curr. Med. Chem., 2007, 14, 1829 CrossRef CAS.
- (a) L. L. Fershtat, M. A. Epishina, A. S. Kulikov, M. I. Struchkova and N. N. Makhova, Chem. Heterocycl. Compd., 2015, 51, 176 CrossRef CAS PubMed; (b) L. L. Fershtat, M. A. Epishina, A. S. Kulikov and N. N. Makhova, Mendeleev Commun., 2015, 25, 36 CrossRef CAS PubMed.
- For selected examples, see: (a) R. Ferioli, G. C. Folco, C. Ferreti, A. M. Gasco, C. Medana, R. Fruttero, M. Civelli and A. Gasco, Br. J. Pharmacol., 1995, 114, 816 CrossRef CAS PubMed; (b) A. M. Gasco, C. Cena, A. Di Stilo, G. Ermondi, C. Medana and A. Gasco, Helv. Chim. Acta, 1996, 79, 1803 CrossRef CAS PubMed; (c) A. M. Gasco, D. Boschi, A. Di Stilo, C. Medana, A. Gasco, P. A. Martorana and K. Schonafinger, Arzneim. Forsch., 1998, 48, 212 CAS; (d) L. Boiani, G. Aguirre, M. Gonzalez, H. Cerecetto, A. Chidichimo, J. J. Cazzulo, M. Bertinaria and S. Guglielmo, Bioorg. Med. Chem., 2008, 16, 7900 CrossRef CAS PubMed.
- For selected examples on NO-donor furoxan-containing hybrid molecules, see: (a) C. Cena, M. L. Lolli, L. Lazzarato, E. Guaita, G. Morini, G. Coruzzi, S. P. McElroy, I. L. Megson, R. Fruttero and A. Gasco, J. Med. Chem., 2003, 46, 747 CrossRef CAS PubMed; (b) M. F. Buonsanti, M. Bertinaria, A. Di Stilo, C. Cena, R. Fruttero and A. Gasco, J. Med. Chem., 2007, 50, 5003 CrossRef CAS PubMed; (c) K. Chegaev, C. Cena, M. Giorgis, B. Rolando, P. Tosco, M. Bertinaria, R. Fruttero, P.-A. Carrupt and A. Gasco, J. Med. Chem., 2009, 52, 574 CrossRef CAS PubMed; (d) L. Lazzarato, C. Cena, B. Rolando, E. Marini, M. L. Lolli, S. Guglielmo, E. Guaita, G. Morini, G. Coruzzi, R. Fruttero and A. Gasco, Bioorg. Med. Chem., 2011, 19, 5852 CrossRef CAS PubMed; (e) J. L. Dos Santos, C. Lanaro, R. C. Chelucci, S. Gambero, P. L. Bosquesi, J. S. Reis, L. M. Lima, H. Cerecetto, M. Gonzalez, F. F. Costa and M. C. Chung, J. Med. Chem., 2012, 55, 7583 CrossRef CAS PubMed; (f) Y. Tamboli, L. Lazzarato, E. Marini, S. Guglielmo, M. Novelli, P. Beffy, P. Masiello, R. Fruttero and A. Gasco, Bioorg. Med. Chem. Lett., 2012, 22, 3810 CrossRef CAS PubMed; (g) I. T. Schiefer, L. VandeVrede, M. Fa', O. Arancio and G. R. J. Thatcher, J. Med. Chem., 2012, 55, 3076 CrossRef CAS PubMed; (h) E. Borretto, L. Lazzarato, F. Spallotta, C. Cencioni, Yu. D'Alessandra, C. Gaetano, R. Fruttero and A. Gasco, ACS Med. Chem. Lett., 2013, 4, 994 CrossRef CAS PubMed; (i) S. Guglielmo, D. Cortese, F. Vottero, B. Rolando, V. P. Kommer, D. L. Williams, R. Fruttero and A. Gasco, Eur. J. Med. Chem., 2014, 84, 135 CrossRef CAS PubMed.
- For selected examples, see: (a) J. P. Agrawal and R. D. Hodgson, in Organic Chemistry of Explosives, Wiley, Chichester, 2007, pp. 302–307 Search PubMed; (b) V. I. Pepekin, B. L. Korsunskii and Yu. N. Matyushin, Combust., Explos. Shock Waves (Engl. Transl.), 2008, 44, 110 CrossRef; (c) A. I. Stepanov, D. V. Dashko and A. A. Astrat'ev, Cent. Eur. J. Energ. Mater., 2012, 9, 329 CAS; (d) L. Liang, K. Wang, C. Bian, L. Ling and Z. Zhou, Chem.–Eur. J., 2013, 19, 14902 CrossRef CAS PubMed; (e) A. O. Chizhov, N. N. Makhova, I. V. Kuchurov, A. B. Sheremetev and S. G. Zlotin, Mendeleev Commun., 2014, 24, 165 CrossRef CAS PubMed; (f) D. Fischer, T. M. Klapötke and J. Stierstorfer, Eur. J. Inorg. Chem., 2014, 5808 CrossRef CAS PubMed.
- For selected examples on 1,2,4-oxadiazole rearrangements, see: (a) N. Vivona, S. Buscemi, V. Frenna and G. Cusmano, Adv. Heterocycl. Chem., 1993, 56, 49 CAS; (b) S. Buscemi, V. Frenna, A. Pace, N. Vivona, B. Cosimelli and D. Spinelli, Eur. J. Org. Chem., 2002, 1417 CrossRef CAS; (c) N. N. Makhova, I. V. Ovchinnikov, A. S. Kulikov, S. I. Molotov and E. L. Baryshnikova, Pure Appl. Chem., 2004, 76, 1691 CrossRef CAS; (d) A. Pace, I. Pibiri, A. Palumbo Piccionello, S. Buscemi, N. Vivona and G. Barone, J. Org. Chem., 2007, 72, 7656 CrossRef CAS PubMed; (e) A. Palumbo Piccionello, A. Pace, S. Buscemi, N. Vivona and M. Pani, Tetrahedron, 2008, 64, 4004 CrossRef CAS PubMed; (f) A. Pace, P. Pierro, S. Buscemi, N. Vivona and G. Barone, J. Org. Chem., 2009, 74, 351 CrossRef CAS PubMed; (g) A. Pace and P. Pierro, Org. Biomol. Chem., 2009, 7, 4337 RSC; (h) A. P. Piccionello, A. Guarcello, A. Pace and S. Buscemi, Eur. J. Org. Chem., 2013, 1986 CrossRef PubMed.
- (a) I. Vega, W. Morris and N. D'Accorso, React. Funct. Polym., 2006, 66, 1609 CrossRef CAS PubMed; (b) I. Vega, L. Sanchez and N. D'Accorso, J. Heterocycl. Chem., 2007, 44, 389 CrossRef CAS PubMed; (c) R. Agneeswari, V. Tamilavan, M. Song, J.-W. Kang, S.-H. Jin and M. H. Hyun, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2131 CrossRef CAS PubMed; (d) Q. Li, L.-S. Cui, C. Zhong, Z.-Q. Jiang and L.-S. Liao, Org. Lett., 2014, 16, 1622 CrossRef CAS PubMed; (e) D. Ko, H. A. Patel and C. T. Yavuz, Chem. Commun., 2015, 51, 2915 RSC.
- (a) K. Luthman, S. Borg and U. Hacksell, Methods Mol. Med., 1999, 23, 1 CAS; (b) J. C. Jochims, in Comprehensive Heterocyclic Chemistry, ed. A. R.Katritzky, C. W.Rees and E. F. V.Scriven, Pergamon Press, Oxford, 2nd edn, 1996, vol. 4, ch. 4.04, pp. 179–228 Search PubMed; (c) K. Hemming, in Comprehensive Heterocyclic Chemistry, ed. A. R.Katritzky, C. A. Ramsden, E. F. V.Scriven and R. J. K. Taylor, Elsevier, Amsterdam, 3rd edn, 2008, vol. 5, ch. 5.04, pp. 243–314 Search PubMed.
- J. Xu, L. Wei, R. Mathvink, J. He, Y.-J. Park, H. He, B. Leiting, K. A. Lyons, F. Marsilio, R. A. Patel, J. K. Wu, N. A. Thornberry and A. E. Weber, Bioorg. Med. Chem. Lett., 2005, 15, 2533 CrossRef CAS PubMed.
- (a) M. A. Weidner-Wells, T. C. Henninger, S. A. Fraga-Spano, C. M. Boggs, M. Matheis, D. M. Ritchie, D. C. Argentieri, M. P. Wachter and D. Hlasta, Bioorg. Med. Chem. Lett., 2004, 14, 4307 CrossRef CAS PubMed; (b) N. M. M. Bezerra, S. P. De Oliveira, R. M. Srivastava and J. R. Da Silva, Il Farmaco, 2005, 60, 955 CrossRef CAS PubMed.
- (a) A. G. Tyrkov and L. T. Sukhenko, Pharm. Chem. J., 2004, 38, 376 CrossRef CAS; (b) Y. W. Jo, W. B. Im, J. K. Rhee, M. J. Shim, W. B. Kim and E. C. Choi, Bioorg. Med. Chem., 2004, 12, 5909 CrossRef CAS PubMed; (c) Rakesh, D. Sun, R. B. Lee, R. P. Tangallapally and R. E. Lee, Eur. J. Med. Chem., 2009, 44, 460 CrossRef CAS PubMed; (d) C. G. Fortuna, C. Bonaccorso, A. Bulbarelli, G. Caltabiano, L. Rizzi, L. Goracci, G. Musumarra, A. Pace, A. P. Piccionello, A. Guarcello, P. Pierro, C. E. A. Cocuzza and R. Musumeci, Eur. J. Med. Chem., 2013, 65, 533 CrossRef CAS PubMed.
- (a) H.-Z. Zhang, S. Kasibhatla, J. Kuemmerle, W. Kemnitzer, K. Ollis-Mason, L. Qiu, C. Crogan-Grundy, B. Tseng, J. Drewe and S. X. Cai, J. Med. Chem., 2005, 48, 5215 CrossRef CAS PubMed; (b) K. A. Jessen, N. M. English, J. Y. Wang, S. Maliartchouk, S. P. Archer, L. Qiu, R. Brand, J. Kuemmerle, H.-Z. Zhang, K. Gehlsen, J. Drewe, B. Tseng, S. Xiong Cai and S. Kasibhatla, Mol. Cancer Ther., 2005, 4, 761 CrossRef CAS PubMed; (c) S. Fujii, K. Ohta, T. Goto, H. Kagechika and Y. Endo, Bioorg. Med. Chem., 2009, 17, 344 CrossRef CAS PubMed.
- (a) T. Huhtiniemi, T. Suuronen, V. M. Rinne, C. Wittekindt, M. Lahtela-Kakkonen, E. Jarho, E. A. A. Wallén, A. Salminen, A. Poso and J. Leppanen, J. Med. Chem., 2008, 51, 4377 CrossRef CAS PubMed; (b) M. Koufaki, C. Kiziridi, F. Nikoloudaki and M. N. Alexis, Bioorg. Med. Chem. Lett., 2007, 17, 4223 CrossRef CAS PubMed; (c) S. B. Tiwari and D. V. Kohli, Med. Chem. Res., 2008, 17, 386 CrossRef CAS; (d) M. Ono, M. Haratake, H. Saji and M. Nakayama, Bioorg. Med. Chem., 2008, 16, 6867 CrossRef CAS PubMed.
- (a) P. F. Pagoria and M. X. Zhang, US Pat., 0 263 982, 2013; (b) M. A. Kettner and T. M. Klapoetke, Chem. Commun., 2014, 50, 2268 RSC; (c) M. A. Kettner, K. Karaghiosoff, T. M. Klapoetke, M. Suceska and S. Wunder, Chem.–Eur. J., 2014, 20, 7622 CrossRef CAS PubMed; (d) V. Thottempudi, J. Zhang, C. He and J. M. Shreeve, RSC Adv., 2014, 4, 50361 CAS; (e) M. A. Kettner, T. M. Klapoetke, T. G. Witkowski and F. von Hundling, Chem.–Eur. J., 2015, 21, 4238 CrossRef CAS PubMed.
- V. G. Andrianov, Chem. Heterocycl. Compd., 1997, 33, 973 CrossRef CAS.
- For selected examples, see: (a) L. L. Fershtat, I. V. Ovchinnikov and N. N. Makhova, Tetrahedron Lett., 2014, 55, 2398 CrossRef CAS PubMed; (b) A. Ya Kots, M. A. Grafov, Yu. V. Khropov, V. L. Betin, N. N. Belushkina, O. G. Busygina, M. Yu. Yazykova, I. V. Ovchinnikov, A. S. Kulikov, N. N. Makhova, N. A. Medvedeva, T. V. Bulargina and I. S. Severina, Br. J. Pharmacol., 2000, 129, 1163 CrossRef PubMed; (c) I. V. Ovchinnikov, A. O. Finogenov, M. A. Epishina, Yu. A. Strelenko and N. N. Makhova, Mendeleev Commun., 2009, 19, 217 CrossRef CAS PubMed; (d) A. O. Finogenov, M. A. Epishina, A. S. Kulikov, N. N. Makhova, I. V. Anan'ev and V. A. Tartakovsky, Russ. Chem. Bull., Int. Ed., 2010, 59, 2108 CrossRef CAS; (e) A. O. Finogenov, M. A. Epishina, I. V. Ovchinnikov, A. S. Kulikov, I. V. Anan'ev and N. N. Makhova, Russ. Chem. Bull., Int. Ed., 2011, 60, 339 CrossRef CAS PubMed; (f) N. N. Makhova and A. S. Kulikov, Russ. Chem. Rev., 2013, 82, 1007 CrossRef CAS PubMed; (g) A. O. Finogenov, A. S. Kulikov, M. A. Epishina, I. V. Ovchinnikov, Y. V. Nelyubina and N. N. Makhova, J. Heterocycl. Chem., 2013, 50, 135 CrossRef CAS PubMed; (h) L. L. Fershtat, M. I. Struchkova, A. S. Goloveshkin, I. S. Bushmarinov and N. N. Makhova, Heteroat. Chem., 2014, 25, 226 CrossRef CAS PubMed.
- M. Boiani, H. Cerecetto, M. Gonzalez, M. Risso, C. Olea-Azar, O. E. Piro, E. E. Castellano, A. C. Lopez De Cerain, O. Ezpeleta and A. Monge-Vega, Eur. J. Med. Chem., 2001, 36, 771 CrossRef CAS.
- A. B. Sheremetev and S. M. Konkina, Mendeleev Commun., 2003, 13, 277 CrossRef PubMed.
- (a) A. F. Stepan, C. Subramanyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffmann, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414 CrossRef CAS PubMed; (b) K. W. Gillman, J. E. Starrett Jr., M. F. Parker, K. Xie, J. J. Bronson, L. R. Marcin, K. E. McElhone, C. P. Bergstrom, R. A. Mate, R. Williams, J. E. Meredith Jr., C. R. Burton, D. M. Barten, J. H. Toyn, S. B. Roberts, K. A. Lentz, J. G. Houston, R. Zaczek, C. F. Albright, C. P. Decicco, J. E. Macor and R. E. Olson, ACS Med. Chem. Lett., 2010, 1, 120 CrossRef CAS PubMed; (c) V. G. Andrianov, V. G. Semenikhina and A. V. Eremeev, Chem. Heterocycl. Compd., 1991, 27, 646 CrossRef.
- P. Raboisson, A. Breitholtz-Emanuelsson, H. Dahllöf, L. Edwards, W. L. Heaton, M. Isaac, K. Jarvie, A. Kers, A. B. E. Minidis, A. Nordmark, S. M. Sheehan, A. Slassi, P. Ström, Y. Terelius, D. Wensbo, J. M. Wilson, T. Xin and D. A. McLeod, Bioorg. Med. Chem. Lett., 2012, 22, 6974 CrossRef CAS PubMed.
- Q. Li, L.-S. Cui, C. Zhong, Z.-Q. Jiang and L.-S. Liao, Org. Lett., 2014, 16, 1622 CrossRef CAS PubMed.
- H. L. Yale and E. R. Spitzmiller, J. Heterocycl. Chem., 1978, 15, 1373 CrossRef CAS PubMed.
- K. Lukin and V. Kishoreb, J. Heterocycl. Chem., 2014, 51, 256 CrossRef CAS PubMed.
- Y. Dürüst, H. Karakuş, M. Kaiser and D. Tasdemir, Eur. J. Med. Chem., 2012, 48, 296 CrossRef PubMed.
- D. Conole, T. M. Beck, M. Jay-Smith, M. D. Tingle, C. T. Eason, M. A. Brimble and D. Rennison, Bioorg. Med. Chem., 2014, 22, 2220 CrossRef CAS PubMed.
- V. V. Bakharev, A. A. Gidaspov, E. V. Selezneva, V. E. Parfenov, I. V. Ul'yankina, I. S. Nazarova, Yu. T. Palatova and O. S. El'tsov, Chem. Heterocycl. Compd., 2011, 47, 1258 CrossRef PubMed.
- W. A. El-Sayed, O. M. Ali, M. S. Faheem, I. F. Zied and A. A.-H. Abdel-Rahman, J. Heterocycl. Chem., 2012, 49, 607 CrossRef CAS PubMed.
- Ž. Jakopin, Tetrahedron Lett., 2015, 56, 504 CrossRef PubMed.
- P. V. Fish, G. A. Allan, S. Bailey, J. Blagg, R. Butt, M. G. Collis, D. Greiling, K. James, J. Kendall, A. McElroy, D. McCleverty, C. Reed, R. Webster and G. A. Whitlock, J. Med. Chem., 2007, 50, 3442 CrossRef CAS PubMed.
- (a) A. R. Gangloff, J. Litvak, E. J. Shelton, D. Sperandio, V. R. Wang and K. D. Rice, Tetrahedron Lett., 2001, 42, 1441 CrossRef CAS; (b) T. Nakamura, M. Asano, Y. Sekiguchi, Yu. Mizuno, K. Tamaki, T. Kimura, F. Nara, Yu. Kawase, T. Shimozato, H. Doi, T. Kagari, W. Tomisato, R. Inoue, M. Nagasaki, H. Yuita, K. Oguchi-Oshima, R. Kaneko, N. Watanabe, Y. Abe and T. Nishi, Bioorg. Med. Chem. Lett., 2012, 22, 1788 CrossRef CAS PubMed; (c) P. I. O'Daniel, Z. Peng, H. Pi, S. A. Testero, D. Ding, E. Spink, E. Leemans, M. A. Boudreau, T. Yamaguchi, V. A. Schroeder, W. R. Wolter, L. I. Llarrull, W. Song, E. Lastochkin, M. Kumarasiri, N. T. Antunes, M. Espahbodi, K. Lichtenwalter, M. A. Suckow, S. Vakulenko, S. Mobashery and M. Chang, J. Am. Chem. Soc., 2014, 136, 3664 CrossRef PubMed.
- H. E. Ungnade and L. M. Kissinger, Tetrahedron, 1963, 19(suppl. 1), 143 CrossRef CAS.
- G. Vass, D. Dzsotjan, G. G. Lajgut and T. Pasinszki, Eur. Chem. Bull., 2012, 1, 22 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr, 2008, 64, 112 CrossRef CAS PubMed.
- F. Fang, D.-D. Li, J.-R. Li, J. Sun, Q.-R. Du, H.-B. Gong and H.-L. Zhu, RSC Adv., 2013, 3, 26230 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: All spectroscopic data of compounds. CCDC 1059467. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07295f |
| This journal is © The Royal Society of Chemistry 2015 |