
One-step synthesis of ordered mesoporous CoAl2O4 spinel-based metal oxides for CO2 reforming of CH4†
Leilei Xuab,
Jian Zhanga,
Fagen Wangad,
Kaidi Yuanc,
Liangjun Wangc,
Kai Wube,
Guoqin Xuabd and
Wei Chen*abcd
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore, Singapore. E-mail: phycw@nus.edu.sg; Fax: +65-6777-6126; Tel: +65-6516-2921
bSingapore-Peking University Research Center for a Sustainable Low-Carbon Future, 1 CREATE Way, #15-01, CREATE Tower, Singapore 138602, Singapore
cDepartment of Physics, National University of Singapore, 2 Science Drive 3, 117551, Singapore, Singapore
dNational University of Singapore (Suzhou) Research Institute, Suzhou, China
eCollege of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 14th May 2015
Abstract
A series of ordered mesoporous CoAl2O4 spinel-based metal oxides with different Co content were facilely synthesized via a one-step evaporation-induced self-assembly method and utilized as catalysts for CO2 reforming of CH4. These mesoporous catalysts, which have unique textural properties and outstanding thermal stabilities, exhibited excellent catalytic performances under various reaction conditions. The ordered mesostructures in the catalysts could provide sufficiently accessible Co active sites for gaseous reactants and stabilize Co metallic nanoparticles via confinement effects within the mesoporous frameworks. These mesoporous catalysts exhibited better anti-coke abilities than non-mesoporous catalysts or traditional supported catalysts, thus acting as a promising series of potential catalyst candidates for CO2 reforming of CH4.
1 Introduction
In the past decades, the excessive use of fossil fuels has caused severe environmental problems such as increasing concentrations of greenhouse gases (mainly CO2) in the atmosphere.1,2 Attempts to efficiently reduce CO2 levels have recently attracted worldwide attention. Moreover, CO2 can be considered as a valuable and sustainable carbon source for the synthesis of value-added chemical fuels. Intensive research efforts have been devoted to the development of efficient CO2 reduction and conversion technologies.3,4 Among the strategies proposed for converting CO2 into higher energy intermediates, heterogeneous catalysis-based processes are of great interest for their ease of process scalability.5,6 Among them, the catalytic carbon dioxide reforming of methane (CRM, CH4(g) + CO2(g) = 2CO(g) + 2H2(g), ΔH298 = +247 kJ mol−1) has been considered as one of the most promising technologies for large scale CO2 reduction and utilization. Moreover, under this process, two greenhouse gases (CO2 and CH4) can be simultaneously transformed into the value-added synthesis gas resource (H2 + CO).7–14In comparison with other synthesis gas production processes, such as steam reforming of methane (SRM, CH4 + H2O = CO + 3H2, ΔH = +206 kJ mol−1) and partial oxidation of methane (POM, 2CH4 + O2 = 2CO + 4H2, ΔH = −44 kJ mol−1), the present CRM process can generate syngas with a low H2/CO ratio, i.e., close to unity (1/1), which is highly desirable for downstream chemical processes such as hydroformylation and oxo synthesis.12,15–17 However, the highly endothermic nature of this CRM process requires the reaction to be carried out at high temperatures (>600 °C) to achieve acceptable conversion rates. Under such high temperatures severe thermal sintering of the metallic active sites can result.11,18 The CRM process is often accompanied by undesirable side reactions such as methane decomposition reaction (CH4(g) = C(s) + 2H2(g), ΔH298 = +75 kJ mol−1) and Boudouard reaction (2CO(g) = C(s) + CO2(g), ΔH298 = −172 kJ mol−1). Coke formed in these side reactions can also passivate the active centers.9,12,17,19 Both factors can cause the rapid deactivation of the catalyst, not allowing the CRM process for large-scale industry applications. Therefore, it is highly desirable to rationally design an efficient catalyst with high resistance to thermal sintering and carbon deposition.
It has been widely reported that most of the group VIII transition metals are active towards the CRM reaction.12,20–22 Among these metals, noble metals (such as Pt, Rh, Pd, and Ir) usually have higher catalytic activity and anti-coke capacity than non-precious metals (such as Ni and Co). However, it is more practical to develop Ni- and Co-based catalysts for scalable commercial applications due to their lower cost and inherent availability.23–25 Supported Ni catalysts have been widely investigated because of their high initial activity that is comparable with noble catalysts. However, Ni-based catalysts are prone to carbon deposition over the catalyst surface due to their strong C–H bond cleavage ability, leading to rapid catalyst deactivation and blockage of the fixed bed reactor.7,14,23 It was found that the introduction of Co into a Ni-based catalyst could significantly suppress the carbon deposition during the CRM reaction.7,19,26–28 Recently, it has been revealed that Co showed interesting catalytic performance in the CRM reaction.11,21,29–31
For Co-based catalysts, it is also very challenging to prevent catalyst deactivation derived from thermal sintering of the metallic active sites and carbon deposition.23 Based on thermodynamic calculations, the methane decomposition reaction can only be initiated at temperatures above 553 °C and the Boudouard reaction can take place at temperatures below 700 °C. Therefore, it is impossible to simultaneously inhibit the occurrence of these two carbon deposition reactions under typical CRM reaction conditions.12,32 Moreover, the anti-coke performance of the catalyst is closely related to the size of the Co metallic active sites. Large Co metal ensembles easily stimulate carbon deposition, and therefore the thermal sintering of the metallic active sites plays an important role in carbon deposition during the CRM process.33–36 However, under typical CRM reaction conditions, the sintering of the Co particles can easily take place, making it a big challenge to restrict Co metallic active centers within nanometer size.
Traditionally, small particles can be obtained by the impregnation of a porous support with large surface area in a precursor-containing solution.37,38 However, it is difficult to control metal dispersion because metallic particles have a strong tendency to aggregate at high temperatures.18 Intensive research efforts have been made to confine Co active sites in precursors with defined structures such as spinel (CoAl2O4) and solid solutions (Co–Mg–O).34,39,40 The formation of a strong interaction between Co and the lattice structure can significantly improve the anti-sintering performance of the Co active centers. However, these materials usually could not provide enough accessible surface Co active sites because most of the active metal species are homogeneously dispersed in the bulk.
To address these challenges, a series of ordered mesoporous CoAl2O4 spinel-based metal oxides with different Co content were designed and facilely synthesized via a one-step evaporation-induced self-assembly method. These catalysts possess outstanding structural properties and thermal stability. The Co species were homogeneously dispersed among the mesoporous matrix in the form of CoAl2O4 spinel, and thus the Co nanoparticles could be effectively stabilized via the confinement effect. This effectively suppressed the severe sintering of Co nanoparticles under CRM conditions. Furthermore, these mesoporous catalysts with large surface areas, big pore volume, and uniform pore channels can ensure and provide sufficient Co active sites for the gaseous feedstock. This series of materials is promising as ideal catalysts towards CRM reaction and even other catalytic reactions such as CO2 methanation.
2 Experimental
2.1 The synthesis of the ordered mesoporous CoAl2O4 spinel based metal oxide
Ordered mesoporous CoAl2O4 spinel-based metal oxides with different Co content were synthesized via a one-step evaporation induced self-assemble (EISA) strategy.41,42 In a typical synthesis procedure, approximately 1.0 g (EO)20(PO)70(EO)20 triblock copolymer (Pluronic P123, Mn = 5800, Sigma-Aldrich) was dissolved in 20.0 mL absolute ethanol with vigorous stirring. 1.6 mL of 67.0 wt% concentrated nitric acid, 10 mmol aluminum isopropoxide (>98%, Sigma-Aldrich), and quantitative Co(NO3)2·6H2O (Sigma-Aldrich) were then sequentially added. The pink transparent homogeneous sol was transferred to a Petri dish after stirring for at least 5 h at room temperature, and then dish was placed into a temperature and humidity controlled chamber at 60 °C with 50% relative humidity to undergo solvent evaporation for 48 h. A pink xerogel was obtained after EISA process. Calcination was carried out by slowly increasing temperature from room temperature to 700 °C with 1 °C min−1 ramping rate and maintaining this final temperature for 5 h in air. The final ordered mesoporous materials were labeled as OMA-xCo, where x refers to the molar percentage between Co and Al (x mol% = nCo/nAl × 100%, x = 3, 5, 7, 10, and 15, nAl = 10 mmol).To investigate the role of ordered mesostructures in promoting catalytic properties, CoAl2O4 spinel-based metal oxides without ordered mesostructures were also prepared. The obtained non-mesoporous materials were denoted as NPA-xCo, where the meaning of x was the same as that in OMA-xCo. The method of preparing NPA-xCo was similar to that of OMA-xCo, only without the addition of the Pluronic P123 template.
To compare the catalytic properties of the present OMA-xCo catalysts with a traditional supported catalyst, a Co-based catalyst supported on gamma-alumina (γ-Al2O3) was prepared via an incipient wetness impregnation method using Co(NO3)2·6H2O (Sigma-Aldrich) as the precursor of cobalt. After impregnation, the catalyst precursors were dried in an oven at 60 °C for 24 h. Finally, the catalyst precursors were calcined at 700 °C for 5 h. The obtained catalysts were denoted as x%Co/Al2O3, where x refers to the molar percentage between Co and Al.
2.2 Catalyst characterizations
X-ray diffraction (XRD) patterns were recorded on an X'pert Pro multipurpose diffractometer (PANalytical, Inc., Netherlands) with Ni-filtered Cu Kα radiation (0.15046 nm). The N2 adsorption and desorption analyses were carried out using a NOVA 2200e (Quantachrome, US) at −196 °C. Transmission electron microscopy (TEM), selected area electron diffraction (SAED), and energy-dispersed spectroscopy (EDS) measurements were performed on a JEOL 2010F microscope (Japan) under a working voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) analyses of the catalysts were performed on a VG ESCALAB 210 (Thermo Scientific, US) spectrometer. The XPS data were referenced to the carbon species with C 1s at the binding energy of 284.8 eV. H2 temperature programmed reduction (H2-TPR) measurements for the as-prepared catalysts (100 mg) were performed on a ChemBET Pulsar TPR/TPD (Quantachrome, US) utilizing 5 mol% H2/He steam (70 mL min−1) as a reducing agent and the hydrogen consumption was recorded using a TCD detector. Temperature programmed hydrogenation (TPH) characterizations of the spent catalysts were carried out on the same apparatus as described for H2-TPR. The long-term endurance-tested catalyst (30 mg) was submitted to a heat treatment (10 °C min−1 up to 900 °C) in a gas flow (60 mL min−1) of the 5 mol% H2/He mixture. Thermogravimetric (TG) analysis was performed using a TA instrument 2960 (Discovery, US) from room temperature to 900 °C with a heating rate of 10 °C min−1 under air stream.2.3 Catalytic properties measurement
The reaction was performed in a vertical fixed bed quartz reactor at atmospheric pressure. The temperature of the reaction bed was monitored by a coaxial thermocouple, which was protected by a small quartz thermocouple well. The flow rate of reactant gas was precisely regulated by mass flow controllers (Beijing Sevenstar Electronics Co., Ltd., China). For each test, 100 mg of catalyst was loaded on the quartz fixed-bed reactor. Prior to the reaction, the catalyst was pretreated in situ by reducing in a mixed flow of H2 and N2 (10/20 mL min−1) with a heating rate of 1.5 °C min−1 rate to 800 °C, and maintaining at 800 °C for 120 min. Before introducing the reaction gases (CH4/CO2 = 1, without dilution), the catalyst bed was purged with N2 for half an hour to remove adsorbed hydrogen. The reaction products were analyzed online by a gas chromatograph (Agilent 7890B) equipped with an automatic six-port valve and a TCD detector. A TDX-01 packed column was used for the analysis of gaseous products. The conversions of CO2 and CH4 were calculated based on the balance of carbon. Their formulas are listed as follows: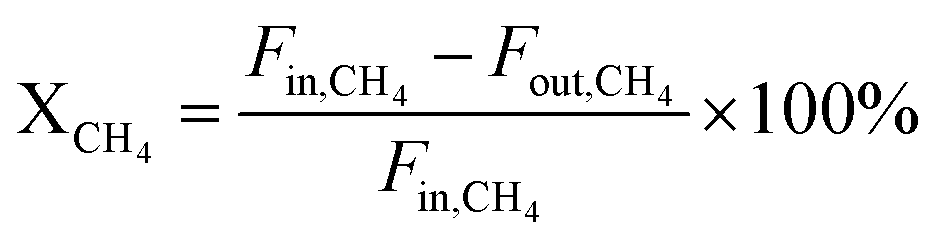
3 Results and discussion
3.1 Characterizations of the fresh catalysts
 | ||
| Fig. 1 (1) Small-angle and (2) wide-angle XRD patterns of the as-synthesized OMA-xCo catalysts: (a) OMA-3Co, (b) OMA-5Co, (c) OMA-7Co, (d) OMA-10Co, and (e) OMA-15Co; (3) XRD patterns of the as-synthesized and (4) as-reduced catalysts prepared by different methods: (a) OMA-10Co, (b) NPA-10Co, and (c) 10%Co/Al2O3. | ||
Comparative studies for OMA-10Co, NPA-10Co, and 10%Co/Al2O3 catalysts prepared with different methods were also carried out and their XRD patterns are displayed in Fig. 1(3). OMA-10Co, synthesized with P123 template, presented weak CoAl2O4 and γ-Al2O3 diffraction peaks. As a comparison, NPA-10Co without any template only displayed apparent CoAl2O4 diffraction peaks that were considerably stronger in intensity than those of OMA-10Co. This suggested that the presence of P123 template was not favorable for phase transformation toward CoAl2O4. The 10%Co/Al2O3 supported catalyst displayed characteristic diffraction peaks for Co3O4 (JCPDS Card no. 74-2120) species and γ-Al2O3 (JCPDS Card no. 10-0425), which could be attributed to the thermal decomposition of the Co(NO3)2·6H2O precursor and γ-Al2O3 support, respectively. Moreover, the positions of the diffraction peaks of CoAl2O4 and Co3O4 crystalline phases almost overlapped, which is consistent with previous studies.43–46 CoAl2O4 and Co3O4 had the same spinel cubic (Fd3m) crystallographic structure with only a slight difference in the lattice constant (a = 8.111 Å for CoAl2O4 and a = 8.0885 Å for Co3O4). Therefore, they displayed similar XRD peak patterns, and the positions of the peaks were nearly same. However, we expected that these catalysts would exhibit different Co 2p XPS and H2-TPR profiles due to the difference in their chemical coordination environments, which will be discussed later.
We also compared the XRD patterns of the as-reduced OMA-10Co, NPA-10Co, and 10%Co/Al2O3 catalysts at 800 °C for 2 h, as exhibited in Fig. 1(4). It could be observed that the intensities of metallic Co diffraction peaks (JCPDS Card no. 15-0806) for as-reduced OMA-10Co and NPA-10Co were weaker than those of 10%Co/Al2O3. For as-reduced OMA-10Co, the diffraction peaks of CoAl2O4 disappeared. As a comparison, the intensities of γ-Al2O3 peaks became stronger after the reduction process. The process of CoAl2O4 reduction (CoAl2O4 + H2 = γ-Al2O3 + H2O) could account for this and the high dispersion of metallic Ni nanoparticles within the mesoporous framework. However, for the as-reduced NPA-10Co, its main crystalline phase remained as CoAl2O4 spinel and its diffraction peaks were considerable stronger than those of metallic Co. This suggests that the reduction of CoAl2O4 spinel species is more difficult. The difference in reducibility between OMA-10Co and NPA-10Co could be attributed to their different structural properties. Compared with NPA-10Co, the larger surface area, bigger pore volume, and unblocked mesopore channels of OMA-10Co made the reduction of CoAl2O4 spinel easier because the H2 molecules has easier access to surface species than bulk material. The pattern of the 10%Co/Al2O3 supported catalyst exhibited obvious metallic Co and γ-Al2O3 diffraction peaks, implying that most Co3O4 species had been reduced.
 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms ((1), (3)) and pore size distribution curves ((2), (4)) of the as-synthesized catalysts. | ||
To investigate the role of the P123 template in the construction of ordered mesoporous channels, comparative analyses between OMA-10Co and NPA-10Co were carried out and the results are displayed in Fig. 2(3) and (4). Unlike OMA-10Co, which has a type IV H1-shaped hysteresis loop and a narrow pore size distribution, NPA-10Co exhibited a type IV H3-shaped hysteresis loop and a wide pore size distribution, suggesting the absence of uniform mesopores. This demonstrated that the P123 template plays a crucial role in constructing ordered mesopores. In addition, the N2 adsorption–desorption analysis result of 10%Co/Al2O3 supported catalysts are also depicted in Fig. 2(3) and (4), showing a type IV H3-shaped hysteresis loop and a wide pore size distribution around 6.0 nm. It is proposed that the mesopores in 10%Co/Al2O3 might be blocked due to the disordered accumulation of γ-Al2O3 support particles. The values of specific surface areas, pore volumes, and average pore diameters of the as-synthesized catalysts are summarized in Table 1. All the OMA-xCo samples calcined at 700 °C exhibited large BET surface areas up to 207.5 m2 g−1, big pore volumes up to 0.31 cm3 g−1, and average pore diameters in the range of 9.8–12.8 nm, which are considerably better than those of NPA-10Co and 10%Co/Al2O3.
| Samples | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) | Isotherm type |
|---|---|---|---|---|
| a AR stands for “as-reduced”.b SP stands for “60 h spent”. | ||||
| OMA-3Co | 148.4 | 0.28 | 9.9 | IV H1 |
| OMA-5Co | 196.5 | 0.27 | 9.8 | IV H1 |
| OMA-7Co | 152.5 | 0.24 | 9.8 | IV H1 |
| OMA-10Co | 207.6 | 0.31 | 9.8 | IV H1 |
| 10Co/Al2O3 | 99.3 | 0.17 | 6.0 | IV H3 |
| NPA-10Co | 85.6 | 0.08 | — | IV H3 |
| OMA-15Co | 150.4 | 0.25 | 12.8 | IV H1 |
| ARa-OMA-3Co | 107.7 | 0.27 | 9.8 | IV H1 |
| AR-OMA-5Co | 124.1 | 0.28 | 9.8 | IV H1 |
| AR-OMA-7Co | 87.2 | 0.19 | 9.9 | IV H1 |
| AR-OMA-10Co | 133.8 | 0.25 | 9.9 | IV H1 |
| AR-10Co/Al2O3 | 96.9 | 0.18 | 6.7 | IV H3 |
| AR-NPA-10Co | 77.5 | 0.05 | — | IV H3 |
| AR-OMA-15Co | 89.2 | 0.19 | 12.6 | IV H1 |
| SP-OMA-7Co | 125.6 | 0.18 | 8.1 | IV H1 |
| SPb-OMA-10Co | 119.8 | 0.15 | 8.0 | IV H1 |
| SP-10Co/Al2O3 | 89.2 | 0.16 | 4.3 | IV H3 |
| SP-NPA-10Co | 61.4 | 0.09 | — | IV H3 |
To further verify the thermal stability of the ordered mesostructures of the OMA-xCo catalysts, the nitrogen adsorption–desorption analyses of the as-reduced OMA-xCo catalysts were carefully carried out and their results are presented in Fig. S1(1) and (2) (ESI†). Identical to their corresponding precursors, the as-reduced samples also fully displayed type IV isotherms with H1-shaped hysteresis loops and narrow pore size distributions around 10.0 nm, demonstrating excellent thermal stabilities. In addition, the isotherms and pore size distribution curves of the as-reduced NPA-10Co and 10%Co/Al2O3 are shown in Fig. S1(3) and (4) (ESI†), respectively. Similar to the as-synthesized samples, they also possessed type IV H3-shaped hysteresis loops and wide pore size distributions. These structural properties of the as-reduced catalysts are also summarized in Table 1. Compared with their corresponding precursors, the specific surface areas and pore volumes suffered some decline after H2 reduction treatment, which might be caused by the shrinkage of the skeleton and phase transformations at high temperature.
 | ||
| Fig. 3 TEM, SAED, and EDS measurements of the as-synthesized OMA-xCo catalysts: (a) OMA-3Co, (b) and (c) OMA-5Co, (d) OMA-7Co, (e) and (g) OMA-10Co, and (f) OMA-15Co. | ||
The TEM characterization of the as-reduced OMA-xCo catalysts was also performed. The as-reduced OMA-3Co, OMA-5Co, and OMA-10Co catalysts were chosen as representatives and their TEM images are displayed in Fig. S2 (ESI†). The alignment of cylindrical pores along the [1 1 0] direction for the as-reduced catalysts was successfully preserved after the reduction process under severe conditions (800 °C for 2 h), demonstrating excellent thermal stability. Furthermore, there was no evident agglomeration of metallic Co within the ordered mesoporous channels, indicating the high dispersion of metallic Co species within the whole mesoporous framework.
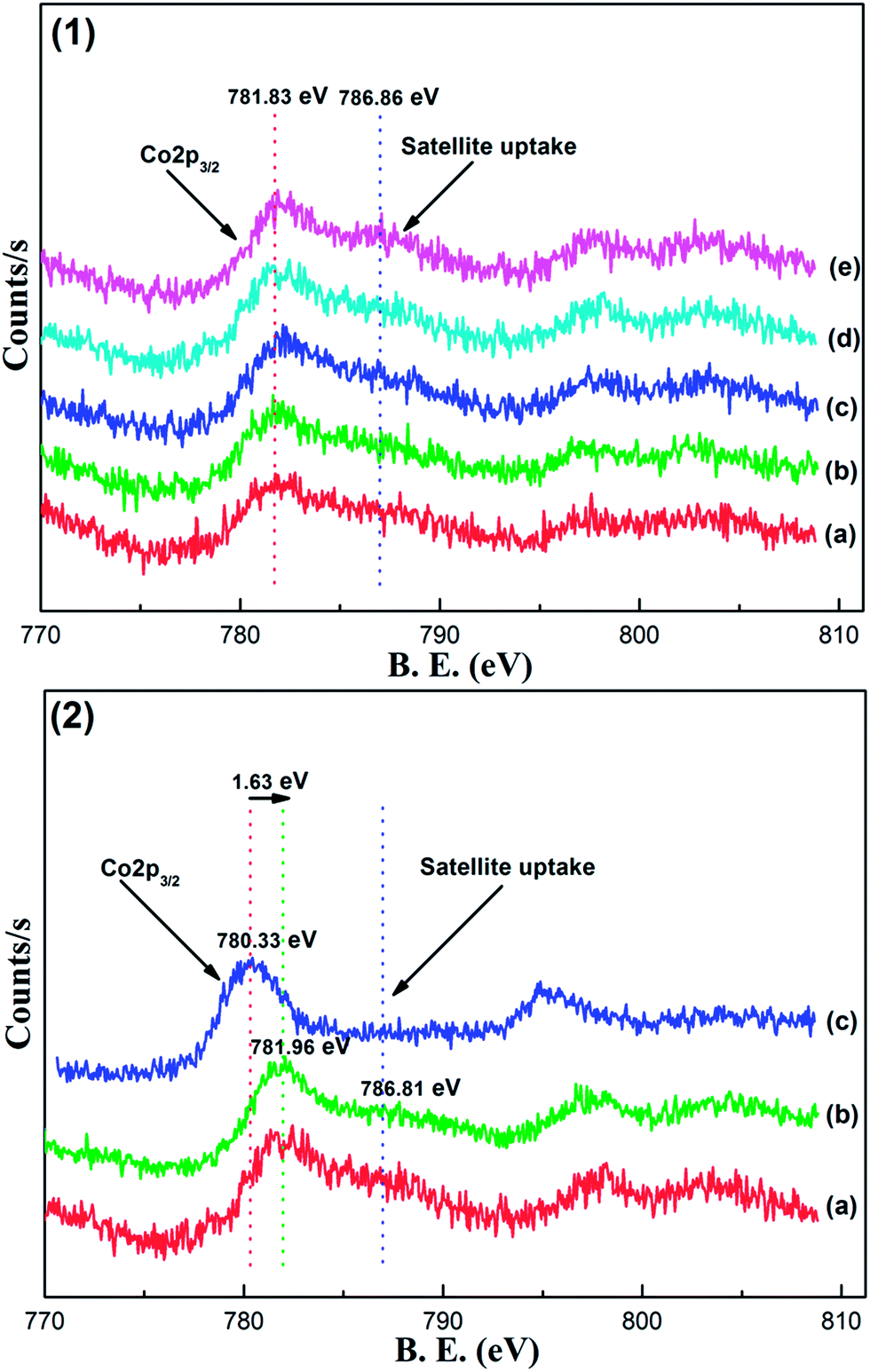 | ||
| Fig. 4 (1) XPS profiles of Co 2p for the as-synthesized OMA-xCo catalysts: (a) OMA-3Co, (b) OMA-5Co, (c) OMA-7Co, (d) OMA-10Co, and (e) OMA-15Co; (2) XPS profiles of Co 2p for the as-synthesized catalysts prepared by different methods: (a) OMA-10Co, (b) NPA-10Co, and (c) 10%Co/Al2O3. | ||
Fig. 4(2) presents the comparative analyses of Co 2p XPS profiles for OMA-10Co, NPA-10Co, and 10%Co/Al2O3 catalysts with the same Co loading. As for NPA-10Co, it exhibited similar patterns to OMA-10Co in both peak position and shape. This suggested that its surface Co species was also present in the form of CoAl2O4 spinel, as verified by XRD analysis in Fig. 1. We propose that during the one-step synthesis process, the precursors of Co and Al species could mix thoroughly and sufficiently react with each other. As a result, Co species could enter into the bulk alumina matrix during the process of calcination, promoting the formation of CoAl2O4 spinel via the following process: CoO + Al2O3 = CoAl2O4. However, for the 10%Co/Al2O3 catalyst, its Co 2p spectrum was considerably different from OMA-10Co in both peak position and shape. In particular, the Co 2p3/2 (780.33 eV) binding energy of 10%Co/Al2O3 was considerably lower than that of OMA-10Co (781.83 eV), which was attributable to a Co3O4 species.45,47,49 In addition, there was no shake-up satellite peak accompanying the Co 2p3/2 peak for 10%Co/Al2O3, which is a typical feature for the CoAl2O4 spinel species.48
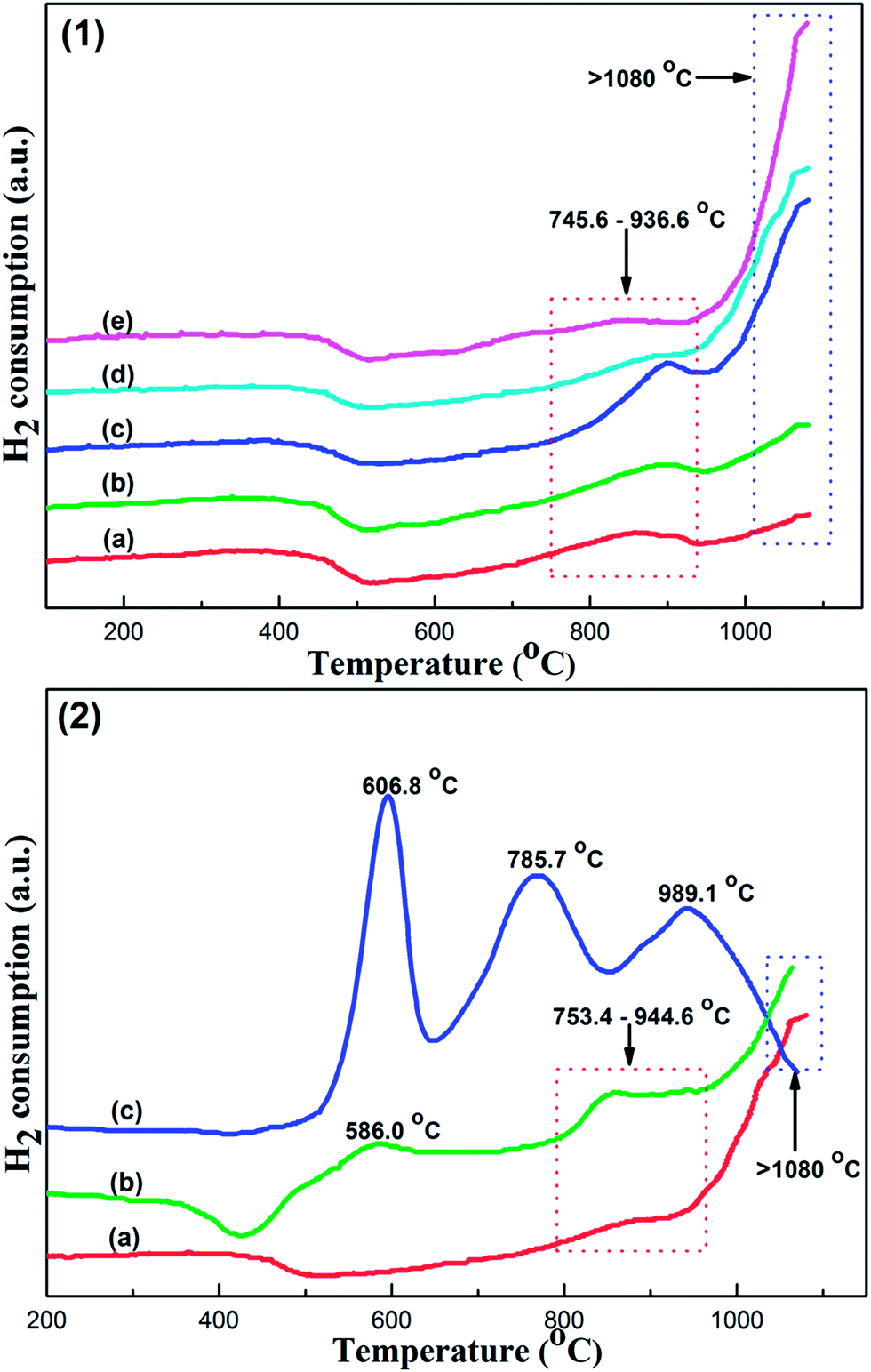 | ||
| Fig. 5 (1) H2-TPR profiles of the OMA-xCo catalysts: (a) OMA-3Co, (b) OMA-5Co, (c) OMA-7Co, (d) OMA-10Co, and (e) OMA-15Co; (2) H2-TPR profiles of the catalysts prepared by different methods: (a) OMA-10Co, (b) NPA-10Co, and (c) 10%Co/Al2O3. | ||
Furthermore, the comparative analysis of H2-TPR profiles for OMA-10Co, NPA-10Co, and 10%Co/Al2O3 catalysts are summarized in Fig. 5(2). For the NPA-10Co catalyst, it presented obvious reduction peaks in a high temperature region similar to OMA-10Co. It displayed high reduction peaks in the high temperature regions, which were also attributed to the reduction of CoAl2O4 spinel species in different coordination environments. In addition, the small uptake peaks in the low temperature region around 586 °C could be attributed to the reduction of Co oxides, which had weak interactions with the bulk catalyst. This might be caused by the absence of a self-assembly step around the P123 template during its synthesis. As a result, the Co species could not be completely transformed into CoAl2O4 and parts of Co species existed as cobalt oxides (CoO or/and Co3O4). In the case of the 10%Co/Al2O3 catalyst, its profile displayed three obvious reduction peaks centered around 607, 786, and 989 °C, which could be attributed to the first step reduction of Co3O4 and subsequent reduction of CoO species. The XRD and XPS analyses had confirmed the presence of Co3O4 crystalline phase over 10%Co/Al2O3 supported catalyst. The reduction peak at 607 °C could be attributed to the first step reduction of Co3O4 (I) (Co3O4 (I) + H2 = 3CoO (I) + H2O), which had weak interactions with the Al2O3 support. The peak centered at 786 °C, however, might stem from both the subsequent reduction of CoO (I) (CoO (I) + H2 = Co + H2O) and the first step reduction of Co3O4 (II) that was strongly interacted with the support. The last peak around 989 °C was attributed to the further reduction of CoO (II) derived from Co3O4 (II), which also had strong interactions with the Al2O3 support.
3.2 Catalytic performances
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL (g−1 h−1), 1 atm) were investigated and the results are summarized in Fig. 6(1) and (2). The conversions of CH4 and CO2 were greatly promoted with an increase in the reaction temperature. Typically, they displayed the highest activity at 800 °C, regardless of Co content due to the intensively endothermic nature of the CRM reaction.12 The relationship between catalytic activity and Co content for OMA-xCo catalysts was also studied. With an increase in Co content, the conversions of CH4 and CO2 rapidly increased in the initial stage and reached a maximum value at 10%Co content for all the temperature stages. This could be attributed to the increasing number of Co active sites, providing the gaseous feedstock with more accessible metallic active sites under similar reduction conditions. However, the higher content of Co up to 15% caused a sharp drop in CH4 and CO2 conversion, which most likely appeared due to the thermal agglomeration of the Co active centers and hence the decrease of active sites.
000 mL (g−1 h−1), 1 atm) were investigated and the results are summarized in Fig. 6(1) and (2). The conversions of CH4 and CO2 were greatly promoted with an increase in the reaction temperature. Typically, they displayed the highest activity at 800 °C, regardless of Co content due to the intensively endothermic nature of the CRM reaction.12 The relationship between catalytic activity and Co content for OMA-xCo catalysts was also studied. With an increase in Co content, the conversions of CH4 and CO2 rapidly increased in the initial stage and reached a maximum value at 10%Co content for all the temperature stages. This could be attributed to the increasing number of Co active sites, providing the gaseous feedstock with more accessible metallic active sites under similar reduction conditions. However, the higher content of Co up to 15% caused a sharp drop in CH4 and CO2 conversion, which most likely appeared due to the thermal agglomeration of the Co active centers and hence the decrease of active sites.
 | ||
| Fig. 6 The curves of (1) CH4 conversion and (2) CO2 conversion versus reaction temperature for OMA-xCo catalysts with different Co content; reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL (g−1 h−1), 1 atm. The curves of (3) CH4 conversion and (4) CO2 conversion versus gas hourly space velocity (GHSV) for OMA-xCo catalysts with different Co content; reaction conditions: CH4/CO2 = 1, 750 °C, 1 atm. | ||
000 mL (g−1 h−1), the conversions of CH4 and CO2 rapidly decreased (e.g. decreased from 88.9% to 60.5% for the CH4 conversion over OMA-10Co) for all the catalysts investigated. It has been proposed that the reduction in the residence time of the reactants on the catalyst surface and the drop in catalyst bed temperature could result in these phenomena.53 However, for the present OMA-xCo catalysts, we propose that the decrease in the residence time over the catalyst surface and the limited number of active sites with respect to the increasing amount of reactants were the main reasons. With the increase of GHSV, the velocity for the gaseous reactants through the catalyst bed became faster and residence time over the catalyst bed became shorter. However, as GHSV increased, the number of reactants moving through the catalyst bed largely increased, making the active sites insufficient for redundant reactants. The effect of Co content on the catalytic activity for OMA-xCo catalysts under different GHSV could also be investigated. Among the OMA-xCo catalysts under given GHSV, OMA-10Co still possessed the highest CH4 and CO2 conversion among all the catalysts studied. It was of great interest to observe that the OMA-10Co always displayed the best catalytic activity among the OMA-xCo catalysts under different GHSVs and at different temperatures. | ||
| Fig. 7 The curves of (1) CH4 conversion and (2) CO2 conversion versus reaction temperature for catalysts prepared with different methods; reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL (g−1 h−1), 1 atm. The curves of (3) CH4 conversion and (4) CO2 conversion versus gas hourly space velocity (GHSV) for catalysts prepared with different methods; reaction conditions: CH4/CO2 = 1, 750 °C, 1 atm. | ||
000 mL (g−1 h−1), CH4/CO2 = 1, 1 atm. As shown in Fig. 8, OMA-10Co and OMA-7Co catalysts, which have ordered mesostructures, exhibited both high catalytic activities and excellent catalytic stabilities during the whole 60 h test. On the other hand, NPA-10Co and 10%Co/Al2O3 samples suffered a gradual decline in CH4 and CO2 conversions (e.g. from 79.9% to 50.6% for CH4 conversion over NPA-10Co). It is well known that developing a catalyst with enhanced stability is significant for the future large-scale industrialization of CRM process. Previous studies have revealed that metallic active centers with smaller sizes possess strengthened capacity of coke suppression, also known as the size effect.33,34 For the OMA-10Co and OMA-7Co catalysts, the growth of the Co particles was effectively suppressed by the confinement effect of the mesoporous framework during the processes of catalyst reduction and CRM reaction, thus leading to good catalytic stability. For NPA-10Co, the absence of ordered mesostructure might cause thermal sintering of the metallic active centers. At the same time, due to the weak interaction between metal and support, the thermal agglomeration of the Co species could take place on the 10%Co/Al2O3 supported catalyst, thus leading to a gradual continuous deactivation. The large metallic clusters facilitated methane decomposition and carbon deposition, hence resulting in the rapid deactivation of the catalyst and large amount of surface coke.33,34 All the H2/CO ratios over these catalysts were less than 1, as shown in Fig. 8(3). This was derived from reverse water gas shift side reaction (RWGS, CO2(g) + H2(g) = H2O(g) + CO(g), ΔH298 = +41 kJ mol−1), which consumes H2 and generates CO.12 The H2/CO ratios over OMA-10Co and OMA-7Co mesoporous catalysts were constant around 0.87 and 0.83, respectively, indicating that equilibrium between the coke deposition reaction and elimination reaction had been achieved.53,54 As a comparison, the H2/CO ratio for the NPA-10Co sample decreased continuously from 0.99 to 0.76, implying that a balance between carbon deposition and coke elimination could not be achieved. The rate of carbon deposition was considerably faster than that of the coke elimination reaction. Therefore, coke deposition over the catalyst surface was very serious and the catalyst subsequently suffered quick deactivation.
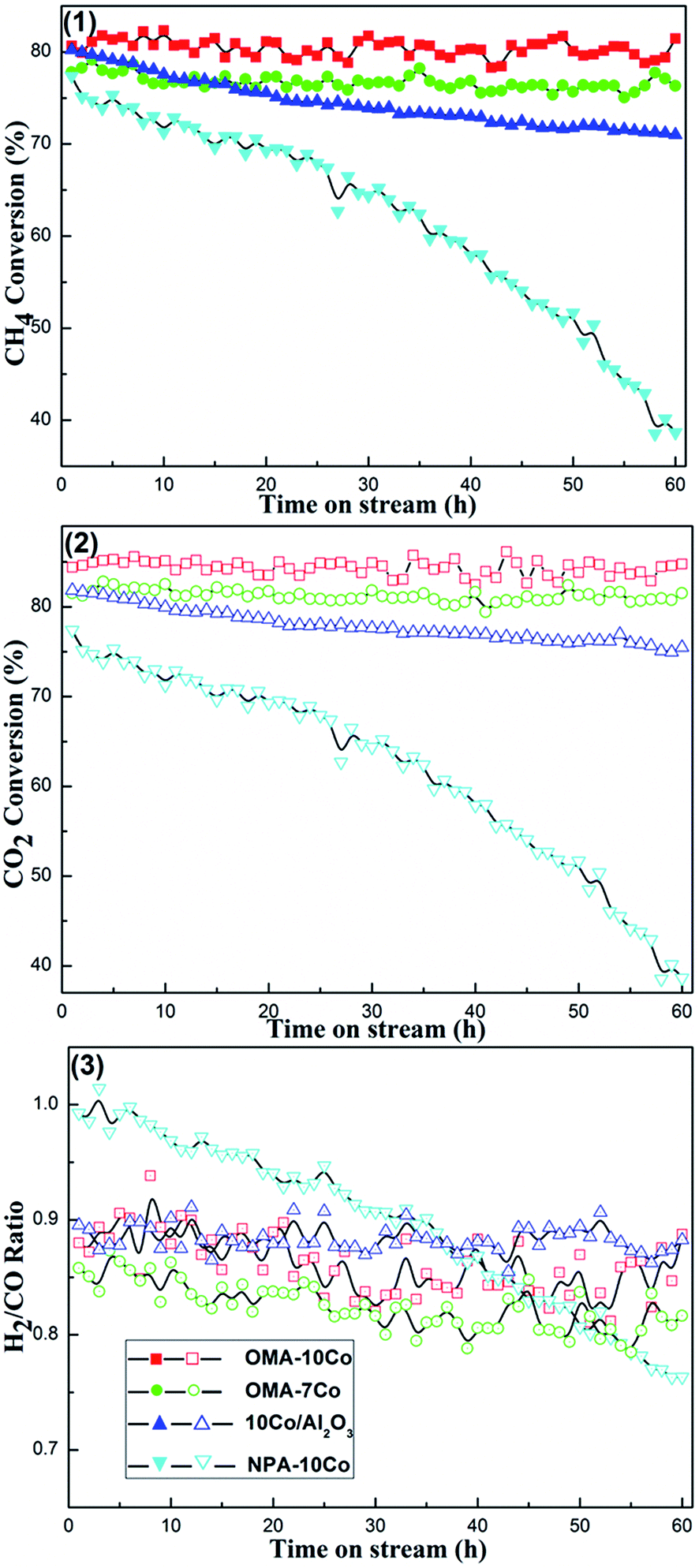 | ||
| Fig. 8 60 h long-term stability tests over different catalysts: (1) CH4 conversion, (2) CO2 conversion, (3) H2/CO ratio; reaction conditions: CH4/CO2 = 1, GHSV = 15000 mL (g−1 h−1), 750 °C, 1 atm. | ||
3.3 Characterization of the spent catalysts
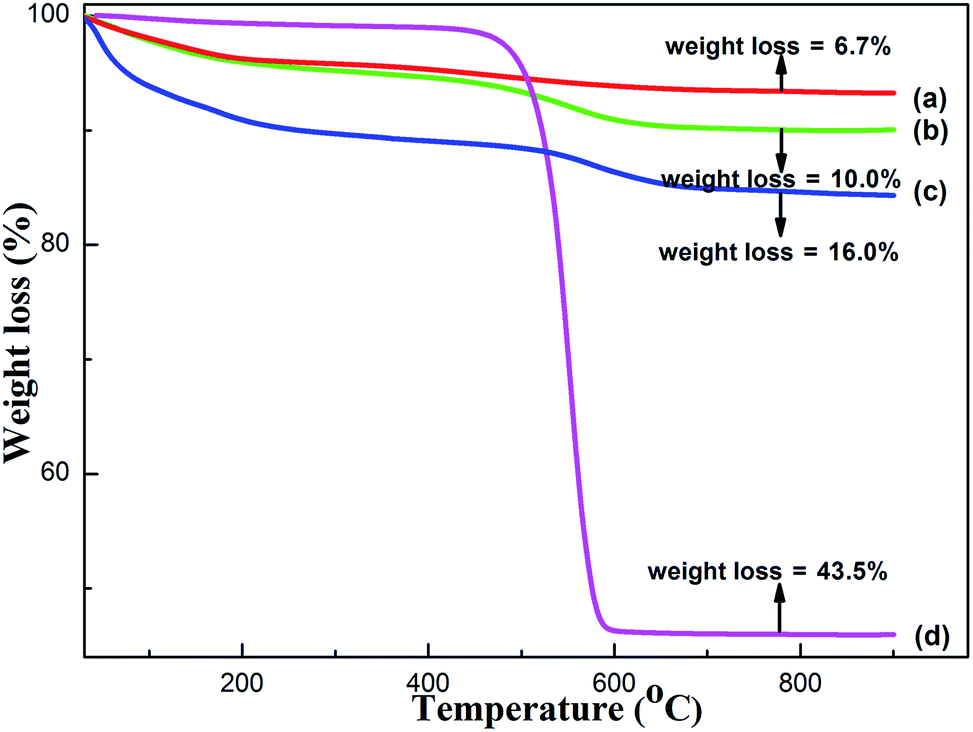 | ||
| Fig. 9 TG curves of the 60 h endurance-tested catalysts: (a) OMA-7Co, (b) OMA-10Co, (c) NPA-10Co, and (d) 10%Co/Al2O3. | ||
 | ||
| Fig. 10 TPH profiles of the 60 h endurance-tested catalysts: (a) OMA-7Co, (b) OMA-10Co, (c) NPA-10Co, and (d) 10%Co/Al2O3. | ||
 | ||
| Fig. 11 TEM and SAED (inset) images of the 60 h endurance-tested catalysts: (a), (b), and (c) OMA-7Co; (d), (e), and (f) OMA-10Co; (g) NPA-10Co; (h) and (i) 10%Co/Al2O3. | ||
4 Conclusion
In conclusion, a series of ordered mesoporous CoAl2O4 spinel-based metal oxides with different Co content were designed and facilely prepared by a one-step evaporation induced self-assemble method for the first time. The obtained materials exhibited excellent structural properties and good thermal stability, and were utilized as catalysts for the CRM reaction. Compared with non-mesoporous and traditional supported catalysts, these mesoporous catalysts exhibited higher catalytic activities and better stabilities. The ordered mesoporous structure could significantly improve catalytic activity and stability by providing sufficiently accessible Co metallic active sites for gaseous feed stocks and stabilizing the Co nanoparticles via the confinement effect of the mesoporous framework during the CRM reaction, simultaneously. In addition, the OMA-xCo catalysts had a strong capacity for coke resistance due to the size effect of the Co nanoparticles. Our results demonstrated that these OMA-xCo materials could be considered as a series of promising catalysts for the CRM reaction.Acknowledgements
The authors acknowledge the financial support from Singapore MOE grant R143-000-542-112, Singapore National Research Foundation CREATE-SPURc program R-143-001-205-592, and Jiangsu Province Government Research Platform Grant and NUSRI Seed Fund.References
- A. Baiker, Appl. Organomet. Chem., 2000, 14, 751–762 CrossRef CAS.
- I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS PubMed.
- K. Mette, S. Kuehl, H. Duedder, K. Kaehler, A. Tarasov, M. Muhler and M. Behrens, ChemCatChem, 2014, 6, 100–104 CrossRef CAS PubMed.
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- R. Schloegl, ChemSusChem, 2010, 3, 209–222 CrossRef CAS PubMed.
- A. T. Ashcroft, A. K. Cheetham, M. L. H. Green and P. D. F. Vernon, Nature, 1991, 352, 225–226 CrossRef CAS PubMed.
- W. Chen, G. Zhao, Q. Xue, L. Chen and Y. Lu, Appl. Catal., B, 2013, 136, 260–268 CrossRef PubMed.
- Z. Liu, J. Zhou, K. Cao, W. Yang, H. Gao, Y. Wang and H. Li, Appl. Catal., B, 2012, 125, 324–330 CrossRef CAS PubMed.
- M. Yu, K. Zhu, Z. Liu, H. Xiao, W. Deng and X. Zhou, Appl. Catal., B, 2014, 148, 177–190 CrossRef PubMed.
- S. Zeng, X. Zhang, X. Fu, L. Zhang, H. Su and H. Pan, Appl. Catal., B, 2013, 136, 308–316 CrossRef PubMed.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev.: Sci. Eng., 1999, 41, 1–42 CAS.
- M. S. Fan, A. Z. Abdullah and S. Bhatia, ChemCatChem, 2009, 1, 192–208 CrossRef CAS PubMed.
- C. J. Liu, J. Ye, J. Jiang and Y. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS PubMed.
- B. Q. Xu and W. M. H. Sachtler, J. Catal., 1998, 180, 194–206 CrossRef CAS.
- J. R. H. Ross, A. N. J. vanKeulen, M. E. S. Hegarty and K. Seshan, Catal. Today, 1996, 30, 193–199 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Catal. Rev.: Sci. Eng., 2002, 44, 423–453 CAS.
- N. Sun, X. Wen, F. Wang, W. Wei and Y. Sun, Energy Environ. Sci., 2010, 3, 366–369 CAS.
- K. Takanabe, K. Nagaoka, K. Nariai and K. Aika, J. Catal., 2005, 232, 268–275 CrossRef CAS PubMed.
- S. Liu, L. Guan, J. Li, N. Zhao, W. Wei and Y. Sun, Fuel, 2008, 87, 2477–2481 CrossRef CAS PubMed.
- P. Ferreira-Aparicio, A. Guerrero-Ruiz and I. Rodriguez-Ramos, Appl. Catal., A, 1998, 170, 177–187 CrossRef CAS.
- J. H. Edwards and A. M. Maitra, Fuel Process. Technol., 1995, 42, 269–289 CrossRef CAS.
- A. W. Budiman, S. H. Song, T. S. Chang, C. H. Shin and M. J. Choi, Catal. Surv. Asia, 2012, 16, 183–197 CrossRef CAS.
- C. Shi and P. Zhang, Appl. Catal., B, 2012, 115, 190–200 CrossRef PubMed.
- X. Y. Quek, D. Liu, W. N. E. Cheo, H. Wang, Y. Chen and Y. Yang, Appl. Catal., B, 2010, 95, 374–382 CrossRef CAS PubMed.
- M. S. Fan, A. Z. Abdullah and S. Bhatia, Appl. Catal., B, 2010, 100, 365–377 CrossRef CAS PubMed.
- J. Zhang, H. Wang and A. K. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS PubMed.
- P. Djinovic, I. G. O. Crnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef CAS PubMed.
- E. Ruckenstein and H. Y. Wang, Appl. Catal., A, 2000, 204, 257–263 CrossRef CAS.
- E. Ruckenstein and H. Y. Wang, J. Catal., 2002, 205, 289–293 CrossRef CAS.
- N. Wang, W. Chu, L. Huang and T. Zhang, J. Nat. Gas Chem., 2010, 19, 117–122 CAS.
- S. B. Wang, G. Q. M. Lu and G. J. Millar, Energy Fuels, 1996, 10, 896–904 CrossRef CAS.
- J. H. Kim, D. J. Suh, T. J. Park and K. L. Kim, Appl. Catal., A, 2000, 197, 191–200 CrossRef CAS.
- H. Y. Wang and E. Ruckenstein, Appl. Catal., A, 2001, 209, 207–215 CrossRef CAS.
- W. Zhu, A. Borjesson and K. Bolton, Carbon, 2010, 48, 470–478 CrossRef CAS PubMed.
- Y. H. Hu, Catal. Today, 2009, 148, 206–211 CrossRef CAS PubMed.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Catal. Rev.: Sci. Eng., 2006, 48, 363–444 CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova, J. L. G. Fierro, C. Sener and T. Dogu, Appl. Catal., B, 2009, 92, 250–261 CrossRef CAS PubMed.
- L. Y. Mo, J. H. Fei, C. J. Huang and X. M. Zheng, J. Mol. Catal. A: Chem., 2003, 193, 177–184 CrossRef CAS.
- L. Y. Mo, X. M. Zheng, C. J. Huang and J. H. Fei, Catal. Lett., 2002, 80, 165–169 CrossRef CAS.
- Q. Yuan, A. X. Yin, C. Luo, L. D. Sun, Y. W. Zhang, W. T. Duan, H. C. Liu and C. H. Yan, J. Am. Chem. Soc., 2008, 130, 3465–3472 CrossRef CAS PubMed.
- S. M. Morris, P. F. Fulvio and M. Jaroniec, J. Am. Chem. Soc., 2008, 130, 15210–15216 CrossRef CAS PubMed.
- H. Y. Wang and E. Ruckenstein, Catal. Lett., 2001, 75, 13–18 CrossRef CAS.
- R. Liu, M. Yang, C. Huang, W. Weng and H. Wan, Chin. J. Catal., 2013, 34, 146–151 CAS.
- N. Srisawad, W. Chaitree, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, J. Nanomater., 2012, 2012, 95 Search PubMed.
- L. Ji, J. Lin and H. C. Zeng, J. Phys. Chem. B, 2000, 104, 1783–1790 CrossRef CAS.
- R. L. Chin and D. M. Hercules, J. Phys. Chem., 1982, 86, 360–367 CrossRef CAS.
- H. Xiong, Y. Zhang, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2005, 231, 145–151 CrossRef CAS PubMed.
- Z. Ferencz, K. Baán, A. Oszkó, Z. Kónya, T. Kecskés and A. Erdőhelyi, Catal. Today, 2014, 228, 123–130 CrossRef CAS PubMed.
- H. F. Xiong, Y. H. Zhang, J. L. Li and Y. Y. Gu, J. Cent. South Univ. Technol., 2004, 11, 414–418 CrossRef CAS PubMed.
- L. Ji, S. Tang, H. C. Zeng, J. Lin and K. L. Tan, Appl. Catal., A, 2001, 207, 247–255 CrossRef CAS.
- J. L. Li, X. D. Zhan, Y. Q. Zhang, G. Jacobs, T. Das and B. H. Davis, Appl. Catal., A, 2002, 228, 203–212 CrossRef CAS.
- J. M. Wei, B. Q. Xu, J. L. Li, Z. X. Cheng and Q. M. Zhu, Appl. Catal., A, 2000, 196, L167–L172 CrossRef CAS.
- L. Xu, H. Song and L. Chou, Catal. Sci. Technol., 2011, 1, 1032–1042 CAS.
- L. Xu, H. Song and L. Chou, Appl. Catal., B, 2011, 108, 177–190 CrossRef PubMed.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar, P. Bai, X. Liu and Z.-F. Yan, Appl. Catal., B, 2008, 77, 346–354 CrossRef CAS PubMed.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z.-F. Yan, J. Nat. Gas Chem., 2008, 17, 278–282 CrossRef CAS.
- J. Guo, H. Lou and X. Zheng, Carbon, 2007, 45, 1314–1321 CrossRef CAS PubMed.
- H. M. Swaan, V. C. H. Kroll, G. A. Martin and C. Mirodatos, Catal. Today, 1994, 21, 571–578 CrossRef CAS.
- Y. G. Chen and J. Ren, Catal. Lett., 1994, 29, 39–48 CrossRef CAS.
- C. Wang, N. Sun, N. Zhao, W. Wei, J. Zhang, T. Zhao, Y. Sun, C. Sun, H. Liu and C. E. Snape, ChemCatChem, 2014, 6, 640–648 CrossRef CAS PubMed.
- H. Liu, C. Guan, X. Li, L. Cheng, J. Zhao, N. Xue and W. Ding, ChemCatChem, 2013, 5, 3904–3909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07249b |
| This journal is © The Royal Society of Chemistry 2015 |