DOI:
10.1039/C5RA07218B
(Paper)
RSC Adv., 2015,
5, 51158-51168
Greener synthetic route for superparamagnetic and luminescent α-Fe2O3 nanoparticles in binary mixtures of ionic liquid and ethylene glycol
Received
21st April 2015
, Accepted 4th June 2015
First published on 5th June 2015
Abstract
α-Fe2O3 nanoparticles have been synthesized by a facile route employing inherently green binary mixtures of ionic liquid (IL), 1-ethyl-3-methyl imidazolium ethylsulphate, [C2mim][C2OSO3] and ethylene glycol, EG. A room temperature grinding of Fe(NO)3·9H2O in the given binary mixture in the presence of NaOH led to the formation of amorphous particles, which on calcination, yielded α-Fe2O3 nanoparticles (NPs). The obtained NPs have been characterized by various state of the art techniques such as X-ray diffraction, Raman spectroscopy, UV-Vis and photoluminescence spectroscopy. The surface and morphological features of NPs have been investigated using scanning and transmission electron microscopy. The prepared NPs have shown weak ferromagnetic character with an essence of superparamagnetism as probed by vibrating sample magnetometry (VSM) and Mössbauer spectroscopy. The composition of the binary mixture of solvent has been found to affect the size, morphology and characteristic properties of the prepared α-Fe2O3 NPs.
1. Introduction
In the field of nanotechnology, iron oxide nanoparticles (NPs) have attracted a lot of attention from the scientific community due to their peculiar properties and potential applications such as in pigments,1 gas sensors,2 field effect transistors,3 photoelectrolysis reactors,4,5 contrast reagents and drug delivery,6 magnetic storage,7 as photoanodes for possible photo-electrochemical cells,8 and in catalysis9 etc. Among various forms of iron oxide NPs, the most common forms, α-Fe2O3 and γ-Fe2O3 NPs are the extensively employed in various technical applications. However, α-Fe2O3 NPs have received significant attention due to their higher stability under ambient conditions, low cost and non-toxic nature.10 To achieve the desired stability of these NPs with increased efficiency for respective applications, extensive efforts have been made for controlling the size and morphology of α-Fe2O3 NPs. Numerous reports are available in literature regarding the synthesis of α-Fe2O3 NPs with distinct shapes such as flower shaped α-Fe2O3 with high surface area and showing adsorption property of heavy metal ions,11 nanotubes and nanorods with a diameter of 30–50 nm, polyhedral, plate, disc and needle shaped morphology, which showed shape dependent magnetic properties,12,13 nanodiscs (50–60 nm),14 nanocubes having an average size of 15 nm with shape dependent optical properties15 and many more.
On the other hand, ionic liquids (ILs) are the salts which are liquid at or below 100 °C and possess an array of unique physico-chemical properties such as negligible vapor pressure, high solvating ability, large electrochemical window, high ionic conductivity and high decomposition temperature.16,17 Owing to their properties, ILs are attracting great interest from researchers around globe for diverse scientific processes. One such interest is to utilize ILs as solvents and soft templates for the preparation of a variety of nano-structured materials.18–22 There are many reports regarding the preparation of hematite NPs employing ILs using a variety of processes. Zheng et al.,23 have synthesized α-Fe2O3 NPs of varying size and morphology via IL assisted hydrothermal route, possessing a weak ferromagnetic character with a saturation magnetization less than 1 emu g−1. Kim et al., have synthesized superparamagnetic α-Fe2O3 NPs by thermal decomposition of Fe(CO)5 in an IL, [C8mim][BF4]–DMF mixture solvent system.24 He et al., have used hydrophobic IL comprising of iron containing ions for direct synthesis of α-Fe2O3 nanocubes by hydrothermal method and investigated their application towards photo-electrochemical sensing of glucose.25 Zheng et al., have reported the ionothermal synthesis α-Fe2O3 nanoplates possessing a weak ferromagnetic character26 and in another report, porous plate like α-Fe2O3 mesocrystals have been prepared by a controlled solvent evaporation process in the presence of IL, [C4mim][Cl].27 Similarly, photocatalytically active α-Fe2O3 hollow microspheres have been synthesized from iron containing IL, [C8mim][FeCl4] under solvothermal condition.28
As discussed above, till date, a variety of effective methods have been tested for the preparation of α-Fe2O3 NPs including conventional as well as IL assisted methods. Most of these methods require expensive apparatus, multistep procedures, a large amount of energy as well as time and a variety of organic solvents. Therefore the need of hour is to prepare nanomaterials conveniently and environmentally benign methods. Hydrolysis stable ILs are the best choice as relatively greener solvents. Further, ILs possesses low surface tension which can favor high nucleation.29 Besides this, the electronic as well as stearic stabilization, extended hydrogen bonded network and high directional polarizability in ILs leads to extended ordering of nanoscale structures while preventing the aggregation of NPs.30 Further the fine tuning of the characteristic properties of ILs by judicious choice of respective ions of IL adds to the uniqueness of these solvents. On the other hand, EG can also be considered as a relatively greener solvent as compared to that of volatile organic solvents at least in terms of non-volatility of EG at room temperature. Therefore, the binary mixtures of ILs with other solvents such as water or EG are expected to provide unique properties towards self-assembly of amphiphiles and synthesis of nanomaterials, which are not available either in IL or other solvents in bulk.31,32 In this regard, a few reports have been published concerned with the self-assembly of amphiphiles in binary mixture of IL, 1-ethyl-3-methyl imidazolium ethylsulphate, [C2mim][C2OSO3] and water where composition dependent behavior has been observed.
Herein, we report a simple and facile method where simple grinding of metal salt in binary mixture of an IL, 1-ethyl-3-methylimidazolium ethylsulphate, [C2mim][C2OSO3] and ethylene glycol, EG as a solvent yields α-Fe2O3 NPs. The luminescent and ferromagnetic nature of formed α-Fe2O3 NPs with an essence of superparamagnetism have been found to be dependent on the relative amount of IL and EG in their binary mixture. The IL and EG acts as solvent as well as templating agent which prevents the agglomeration of prepared NPs. The effect of composition of IL and EG binary mixture on various physico-chemical properties such as structural, optical and magnetic properties of α-Fe2O3 NPs have been discussed in detail. The novelty of the work lies in the fact that there is no report in literature for the synthesis of NPs, specifically α-Fe2O3 utilizing inherently greener binary mixtures of [C2mim][C2OSO3] and EG where composition dependent behavior towards characteristic properties of α-Fe2O3 is expected. Further the low temperature route without the use of any sophisticated instrument adds to the usefulness of the present work. The α-Fe2O3 NPs thus obtained exhibit high saturation magnetization and negligible coercivity, thus showing superparamagnetism along with luminescence which can find place in diverse biomedical applications.
2. Experimental section
2.1. Synthesis of α-Fe2O3 nanoparticles
Ferric nitrate nonahydrate (Fe(NO)3·9H2O), ethylene glycol (EG), 1-ethyl-3-methyl imidazolium ethylsulphate (IL) and NaOH were purchased from Sigma with purity >98%. In a typical synthesis, 2 mmol of Fe(NO)3·9H2O and different compositions of binary mixture of 1-ethyl-3-methylimidazolium ethylsulphate (IL) and ethylene glycol (EG) were ground for 20 min in mortar to form a viscous liquid. Further 6 mmol of NaOH was added and the mixture was ground for another 20 min which transformed to a thick paste. The thick paste was heated at 90 °C for 24 h to ensure the completeness of reaction. The procedure of synthesis is shown schematically in Scheme 1. The obtained products from different compositions of binary mixture was dried in air after being washed by water and then alcohol which was further heated at 500 °C for 4 h to remove the organic part and to increase the crystallinity of NPs. The obtained products have been coded as αFe-0, αFe-25, αFe-50, αFe-75, αFe-100 where the number denotes the percentage of IL in IL–EG binary mixture.
 |
| | Scheme 1 Stepwise synthesis of α-Fe2O3 NPs in binary mixture of IL and EG. | |
2.2. Characterization
X-ray diffractograms of the prepared nanoparticles (NPs) for phase analysis were recorded using a Rigaku Xpert Pro X-ray diffractometer provided with Cu Kα radiation (1.541 Å) in the 2θ range of 20°–80° at a step size of 0.02°. The average particle size was calculated based on XRD patterns using Scherrer's formula. Raman spectra were obtained for the prepared samples by placing the sample on a glass slide, using Renishaw Raman microscope equipped with 488 nm Ar-ion laser in the range of 100–2000 cm−1. UV-Vis spectra were recorded on UV-spectrophotometer (UV-1800 SHIMADZU). Photoluminescence spectra were recorded on a Perkin Elmer spectrometer using the excitation wavelength of 390 and 450 nm at a slit width of 2.5 nm for both excitation and emission. Both UV-visible and photoluminescence measurements were carried out in dispersed form using ethanol as the solvent. The Mössbauer spectra of the NPs were measured using a conventional constant acceleration drive Mössbauer spectrometer with 57Co γ-ray source of 25 mCi embedded in rhodium matrix. The isomer shift values are reported with respect to pure iron absorber. Magnetic studies were carried out at room temperature in the applied magnetic field of −20 to +20 kOe using Microsense EV-90 vibrating sample magnetometer. The surface morphology of the NPs was investigated by scanning electron microscopy (SEM) using Zeiss Ultra 55-Limited edition scanning electron microscope. Transmission electron microscope (TEM) images were recorded using JEM-2100 transmission electron microscope at a working voltage of 200 kV. The NPs were dispersed into ethanol using ultrasonicator for sample preparation for SEM and TEM measurements. A drop of dispersion was placed on the carbon coated grid (300 Mesh) and excess solution was blotted off. The samples were dried at room temperature for 24 hours before the measurement.
3. Result and discussion
We herein report a convenient method for the synthesis of α-Fe2O3 nanoparticles (NPs) by simple grinding protocol. Binary mixtures of an ionic liquid (IL), 1-ethyl-3-methylimidazolium ethylsulfate, [C2mim][C2OSO3] and ethylene glycol (EG) in different mixing ratios have been used as a solvent for precipitation induced formation of α-Fe2O3 NPs. The composition of binary mixture of IL and EG played a vital role in controlling the size and thus in modulation of its various physico-chemical properties. In pure IL, the size of NPs is small with low crystallinity, whereas in pure ethylene glycol (EG) crystallite size becomes relatively very large. However, the binary mixture of IL and EG played very important role in confining the size of NPs and modifying the optical and magnetic properties of prepared NPs. All the results obtained have been correlated with the nature of the solvent used for the preparation of NPs and discussed one by one.
3.1. Structural studies
The XRD patterns of obtained NPs by using different composition of ionic liquid (IL) and ethylene glycol (EG) are shown in Fig. 1(a). The diffraction peaks observed for obtained products matches very well with the hematite (JCPDS card no. 72-0469) indicating the formation of rhombohedral α-Fe2O3 phase (a = 5.038 Å, c = 13.772 Å). From the XRD studies, it has been observed that the with the increase in content of IL in IL–EG binary solvent, the crystallinity of obtained NPs decreases as indicated by increase in half line width of the respected diffraction peaks. Here the coordinating nature of the solvent along with their dielectric constant is expected to play an important role in deciding the degree of crystallization and size of the crystallites of NPs. Further, the small peak (012) is quite clear in samples prepared at lower content of IL, however, it seems to be absent in the samples prepared at quite higher concentration of IL. The reason behind such behavior is supposed to be the decrease in crystallinity of the obtained NPs particles with increase in content of IL in IL–EG binary mixture. Another reason for the absence of (012) peak in αFe-75 and αFe-100 may be the stronger interactions of growing NPs with IL, where the growth of NPs along (012) direction is restricted. The average crystallite size of prepared NPs has been obtained using Scherrer's equation as follows:
D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ |
where D is the particle size perpendicular to the normal line of (hkl) plane, K is a constant (0.9), β is the full width at half-maximum of the (hkl) diffraction peak, θ is the Bragg angle, and λ is the wave-length of X-ray. The variation of obtained size of crystallites as a function of content of IL in IL–EG binary mixture is shown in Fig. 1(b). The size of crystallites for all the prepared NPs lies in the range of 12–47 nm, depending upon the composition of binary mixture of IL and EG. Similar to that of degree of crystallinity, the crystallite size has been found to decrease with increase in content of IL in the IL–EG binary solvent. The crystallite size decreases sharply while moving from αFe-0 to αFe-25 which can be related to change in characteristic properties of the binary solvent. The binary solvent mixtures comprising ILs and other solvent shows completely different physicochemical properties which are not present in the either of the pure solvents.31,32 In the IL–EG binary solvent, there exists the dominance of dispersion forces over specific interactions, which increases with increase in content of IL leading to decrease in interactions between constituent ions of IL and EG.31,32 This results in enhanced coordinating ability of constituent ions of IL as well as of EG towards different reaction species. During the preparation of α-Fe2O3 NPs, Fe(OH)3 or FeOOH forms as an intermediate after the addition of NaOH as per the following equation:
 |
| | Fig. 1 (a) XRD spectra of α-Fe2O3 NPs synthesized in binary mixture of IL and EG; and (b) variation of particle size as a function of composition of IL–EG binary mixture. | |
Both the EG and constituent ions of IL are expected to interact with Fe(OH)3 and the growing α-Fe2O3 NPs. In the EG–IL binary mixture, at lower content of IL (25 vol%), the ions of IL and EG interacts very strongly with each other.31,32 As a consequence of this, constituent ions of IL and EG interact (H-bonding and ion-dipole interactions) feebly with the Fe(OH)3 leading to their less adsorption on its surface which results in unrestricted growth of NPs. This thereby exerts very less effect on degree of crystallinity during the formation of α-Fe2O3. Further, as the solvent coordinates weekly with the Fe(OH)3, it does not assert the growth of NPs leading to larger size of crystallites at lower content of IL in binary mixture of IL and EG. However, with an increase in content of IL, the interactions between constituent ions of IL and Fe(OH)3 as well as EG and Fe(OH)3 increases at the cost of deceased interactions between IL and EG, where IL acts as a template and thus restricts the growth of crystallites leading to smaller size of NPs. Based on above discussions, a schematic showing the possible mode of interactions between the different components of the reaction mixture is provided as Scheme 2.
 |
| | Scheme 2 Proposed mechanism for the formation of α-Fe2O3 NPs synthesized in different compositions of IL and EG. | |
The room temperature Raman spectra of α-Fe2O3 NPs are shown in Fig. 2. The Raman bands of obtained NPs have been found to be broadened due to which exact band position could not be located. Further the broadening of the Raman peaks is a common feature of NPs.33 The three Raman modes for α-Fe2O3 NPs around at 216 (A1g), 283 (Eg), and 398 (Eg) cm−1 has been observed, which is in agreement with the literature values.34,35 All the Raman bands observed for α-Fe2O3 NPs shifts towards lower wavenumber with increase in content of IL in IL–EG binary mixture with relatively greater shifting in Raman bands present around 283 and 398 cm−1. Apart from decrease in size of α-Fe2O3 with increase in content of IL, this shift may be due to interaction of constituent ions of IL interacting with α-Fe2O3 in their crystal lattice. This assumption is well supported by decrease in crystallinity of α-Fe2O3 NPs with increase in IL content. The sample αFe-100 shows very broadened and less intense Raman bands owing to is low crystallinity and smallest size. Thus from Raman and XRD studies, we have concluded that both IL and EG helps in controlling the size and crystallinity of NPs.
 |
| | Fig. 2 Raman spectra of α-Fe2O3 NPs synthesized in binary mixture of IL and EG. | |
3.2. Morphological studies
Morphology and size of the NPs have been characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) techniques and are shown in Fig. 3 and 4, respectively. SEM images indicate that the prepared α-Fe2O3 NPs are roughly spherical in shape. We have not observed any distinct morphological changes for the synthesized NPs from SEM measurements. This indicates that the presence of IL could only affect the size and degree of crystallinity without affecting the morphology of the NPs. Although, the IL 1-ethyl-3-methylimidazolium ethylsulfate has a capability to coordinate with the Fe(OH)3 as an intermediate as well as with the growing NPs, however this small chain IL lacks the amphiphilic character required to control the shape of the NPs by acting as structure directing template. It can be seen from the SEM images that with increase in content of IL in binary solvent, the size of the NPs decreases corroborating with the XRD results. The NPs obtained from lower content of IL (αFe-0 to αFe-50) are appear to be more disperse than those obtained from higher content of IL (αFe-75 and αFe-100) which shows some aggregation with decrease in particle size and this type of behavior also seen in TEM studies. The IL exhibits lower surface tension as compared to that of water, thus resulting in high nucleation rate than the growth rate, which helps in formation of relatively smaller NPs.29,36 Here the role of polarity and viscosity of solvent in controlling the size of NPs is as important as the coordination of constituents of binary solvent with Fe(OH)3 in deciding the size and crystallinity of NPs. The polarity and viscosity of IL–EG binary mixture decreases and increases respectively, with increase in content of IL. A decrease in polarity is expected to enhance the aggregation, whereas an increase in viscosity is assumed to decrease the extent of aggregation of NPs. A decrease in size of the NPs with increase in content of IL thus indicates that the size of the NPs are being controlled by the viscosity factor as a dominating one along with the enhanced coordination of the constituents of binary solvents with the NPs with increase in content of IL.
 |
| | Fig. 3 SEM images of α-Fe2O3 NPs synthesized using different compositions of IL–EG binary mixture. | |
 |
| | Fig. 4 TEM images of α-Fe2O3 NPs synthesized using different compositions of IL and EG (SAED pattern shown in inset). | |
As can be seen from Fig. 4, TEM images strongly support the observation made by SEM measurements. As we move from αFe-0 to αFe-100, a decrease in particle size has been observed in corroboration with the results obtained from XRD and SEM measurements. The αFe-0 NPs have been found to be a mix of near spherical to rectangular in shape (20–40 nm). The NPs obtained for αFe-25 are near spherical in shape (14–19 nm), whereas NPs obtained as αFe-50 (10–15 nm) don't reveal any specific morphology. With further increase in content of IL i.e. for αFe-75, agglomerated NPs with near spherical shape (6–10 nm) have been observed indicating the effect of composition of binary solvent mixture on shape of the NPs. Very surprisingly, in neat IL, the formed NPs (αFe-100) adopted an interconnected network of NPs (7–11 nm) having a hollow space between interconnections. Although the IL is homogeneous macroscopically, however the existence of nano-segregation even in small chain ILs, where polar and non-polar domains are separated microscopically is a well established phenomenon.37 It seems that the IL and the intermediate i.e. Fe(OH)3 forms a complex structure where Fe(OH)3 arranges itself in a way similar to core–shell structure having IL in the core and Fe(OH)3 at the periphery. The hydrogen-bonding interactions between sulfate anion of IL and hydroxyl group of Fe(OH)3 are assumed to be responsible for stabilization of such type of structures. The washing of the final product after calcinations removes the IL trapped inside the network of formed α-Fe2O3 NPs leading to formation of nanoholes. The SAED pattern (as shown in the inset of TEM images) supports the extent of crystallinity of α-Fe2O3 NPs obtained in different binary solvent mixtures. The SAED pattern shows concentric rings along with some spots thus indicating the polycrystalline nature of the prepared samples. However, in case of NPs obtained at high content of IL in binary mixture (αFe-50 to αFe-100), a clear diffraction pattern has not been observed. Further, the reduced number of diffraction rings for the NPs obtained from higher content of IL (αFe-75, αFe-100) may indicates the decrease in crystallinity of obtained NPs and thus can be corroborated with the XRD measurements.
3.3. UV-Vis studies
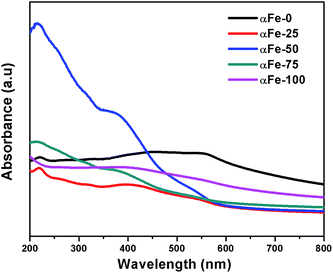
The optical absorption properties of NPs are greatly dependent on particle size and shape and to explain this behavior, UV-Vis spectra have been recorded for the synthesized α-Fe2O3 NPs and are shown in Fig. 5. The synthesized NPs show an absorption band around 210–225 nm region in UV region and a broad band spanning between 400–600 nm in visible region. The band in the visible region seems to be comprised of two sub-bands i.e. a band at 400 and 600 nm. The main contribution of optical absorption spectra of Fe3+ containing substances are d–d transition, ligand to metal charge-transfer transition and the simultaneous excitations of two neighboring Fe3+ cations that are magnetically coupled i.e. pair excitations.38 The variation in absorbance maxima for band around 400 nm is much clearer than that for band around 600 nm when compared for α-Fe2O3 NPs obtained from different binary mixtures. The band at around 400 nm shows a significant blue shift while going form αFe-0 (410 nm) to αFe-50 (375 nm) due to decrease in particle size, whereas surprisingly again a red shift has been observed while going from αFe-50 to αFe-100. This red shift can be explained on the basis of development of surface stress39 due to greater reduction in particle size by increase in the content of IL in binary mixture. The UV spectra reveals that NPs obtained from different IL–EG binary mixtures have different photo-physical properties which can be related to size, crystallinity and state of aggregation in NPs.
 |
| | Fig. 5 UV-Vis spectra of α-Fe2O3 NPs synthesized using different compositions of IL and EG. | |
3.4. Photoluminescence studies
Photoluminescence spectra of prepared α-Fe2O3 NPs are shown in Fig. 6(a) and (b) where strong photoluminescence confirm their emitting nature. In bulk, α-Fe2O3 does not show photoluminescence due to the local forbidden d–d transition, resonant energy transfer between cations and efficient lattice and magnetic relaxations.40 However hematite nanostructures show photoluminescence due to quantum confinement effect, which results in delocalization and quantization of electronic state.41 At an excitation wavelength of 390 nm, the α-Fe2O3 NPs shows an intense emission band around 587 nm and a weak emission band at 525 nm, whereas at excitation wavelength of 450 nm, an emission band around 675 nm has been observed. The origin of luminescence in α-Fe2O3 NPs can be attributed to the decrease in particle size which leads to the reduction in magnetic interactions as compared to that in bulk due to loss of ferromagnetic long range order. The change in the density of electronic population of valence band also enhanced electron–phonon coupling.42–44 The inset of Fig. 6(a) shows the variation in intensity of emission band around 587 nm for NPs prepared in different binary solvents as a function of content of IL in IL–EG binary mixture. The emission intensity increases while going from αFe-0 to αFe-50, while it decreases moving beyond αFe-50 towards αFe-100. At lower content of IL (0–50%), as discussed in earlier sections, constituent ions of IL don't coordinate efficiently with Fe(OH)3 and hence with forming α-Fe2O3 crystallites, thereby exerting no effect on photophysical properties of α-Fe2O3 NPs. The enhancement in emission intensity can be related to dominance of decrease in size of NPs. However at higher content of IL, despite a decrease in size of NPs, the emission intensity decreases. At such high concentrations of IL, there is dominance of increased coordination of constituent ions of IL with forming α-Fe2O3 crystallites leading to surface modification where IL ions may remain coordinated after formation of α-Fe2O3 NPs. The presence of ions of IL may lead to enhanced lattice and magnetic relaxation leading to decrease in emission intensity at higher content of IL. Similar behavior has been observed while analyzing the emission intensity obtained at an excitation wavelength of 450 nm as can be seen from Fig. 6(b). This observation can be correlated with variation in hyperfine field (discussed in Mössbauer section) which first decreases upto αFe-50 and then increases thus results in the variation of emission intensity. This reduction of hyperfine field for αFe-50 as compared to others may leads to enhancement of its emission intensity.44 Further, the emission spectra shown by different NPs are red shifted as compared to that observed for αFe-0. This can be due to surface modification of NPs by the constituent ions of IL.
 |
| | Fig. 6 Photoluminescence spectra of α-Fe2O3 NPs [λexc = (a) 390 nm and (b) 450 nm]. | |
3.5. Mössbauer analysis
Fig. 7 shows the room temperature Mössbauer spectra of α-Fe2O3 NPs obtained from the different compositions of binary mixture of IL and EG at the room temperature. The detailed values of Mössbauer spectral parameters of α-Fe2O3 NPs are summarized in Table 2. For the Mössbauer spectra of αFe-0, a six line hyperfine pattern has been observed similar to that of bulk α-Fe2O3. For αFe-25 also, a sextet pattern has been observed, but with a minor asymmetry as compared to αFe-0. The isomer shift of 0.378 and 0.346 mm s−1 with magnetic hyperfine field of 51.6 and 51.05 T displayed by αFe-0 and αFe-25, respectively, shows a close match with reported values for hematite.45 The presence of central quadrupole doublet along with sextet in the spectra of αFe-50, αFe-75 and αFe-100 can be attributed to the superparamagnetic nature of these NPs.46–48 As the composition of IL above 25%, an appearance of central doublet in the spectra along with less intense sextet which is a typical behavior for magnetic NPs exhibiting superparamagnetic relaxation.49,50 The chemical environment of Fe nuclei may change due to different extent of interaction of growing NPs with IL and EG at different compositions of binary solvent mixture which affects the magnetic properties of prepared NPs. The broadened sextet and duplet in the Mössbauer spectra of αFe-50 has been observed with lower BHF value (47.1 T) which can be attributed to small size and dispersed nature of these particles as indicated more clearly from SEM image. This indicates the reduction in magnetic interactions which are also well supported by increase in absorption and emission intensity of αFe-50 in UV and PL studies, respectively. For sample αFe-75 there is a slight increase in BHF (48.75 T) and for αFe-100 it again increase to somewhat large extent (51.36 T) inspite of its comparable particle size with αFe-75. This behavior is also supported from magnetic studies where increase in MS has been observed from αFe-50 to αFe-100 (Discussed in next section). Such behavior can be explained on the basis that for αFe-100, the IL may interacts more with the surface of the growing particles than inside of growing particles, which interact ferromagnetically due to strong interparticle interaction and thus may be responsible for the increased MS and BHF value. The other possibility may be the lattice strain in small sized particles which is supported by a red shift of low intensity in UV spectra. Therefore it is observed that apart from controlling the size, shape and optical properties of α-Fe2O3 NPs, the composition of binary solvent mixture also controls the magnetic properties of α-Fe2O3 NPs where transformation of ferromagnetic (αFe-0, and αFe-25) to superparamagnetic (αFe-50, αFe-75 and αFe-100) character has been observed.
 |
| | Fig. 7 Mössbauer spectra of α-Fe2O3 NPs synthesized by using different composition of IL–EG binary mixture. | |
Table 1 Physico-chemical parameters of α-Fe2O3 NPs obtained from binary mixture of IL–EG
| Sample |
Average particle size from XRD (nm) |
Particle size from TEM (nm) |
Saturation magnetization (emu g−1) |
Remnant magnetization (emu g−1) |
Coercivity (Oe) |
| αFe-0 |
40.1 |
20–40 |
9.03 |
0.023 |
1.57 |
| αFe-25 |
18.3 |
14–19 |
2.27 |
0.020 |
7.12 |
| αFe-50 |
14.8 |
10–15 |
4.88 |
0.002 |
0.39 |
| αFe-75 |
11.7 |
6–10 |
8.51 |
0.003 |
0.58 |
| αFe-100 |
10.3 |
7–11 |
10.27 |
0.097 |
21.5 |
Table 2 Mössbauer parameters of α-Fe2O3 NPs recorded at room temperature
| Sample |
ISa (mm s−1) |
QS (mm s−1) |
BHF (T) |
Spectral area (%) |
| w.r.t pure iron absorber, S = sextet, D = central doublet. |
| αFe-0 |
0.378 |
— |
51.38 |
100 |
| αFe-25 |
0.346 |
— |
51.05 |
100 |
| αFe-50 |
S = 0.341 |
— |
47.1 |
55 |
| D = 0.372 |
0.808 |
— |
45 |
| αFe-75 |
S = 0.408 |
— |
48.75 |
27 |
| D = 0.383 |
0.786 |
— |
73 |
| αFe-100 |
S = 0.373 |
— |
51.36 |
18 |
| D = 0.364 |
0.707 |
— |
82 |
3.6. Magnetic studies
Magnetic properties of α-Fe2O3 prepared NPs were carried out at room temperature. In general, α-Fe2O3 shows antiferromagnetic nature below Morin temperature (TM = 263 K) and can exhibit weak ferromagnetic behavior between 263 K (TM) and 960 K i.e. at Neel temperature (TN) due to slight canting of spin above TM.51 Fig. 8 shows the magnetic hysteresis loops of α-Fe2O3 NPs, which are indicative of weak ferromagnetic nature of NPs. All types of prepared NPs using different IL–EG binary mixtures show negligible remnant magnetization (Mr) and coercivity (Hc), thus indicating the superparamagnetic nature of NPs (Table 1). The remnant magnetization (Mr) and coercivity (Hc) of obtained NPs are less as compared to that reported in literature52 indicating the presence of superparamagnetism in prepared NPs. The saturation magnetization obtained for synthesized NPs lies in the range of 2.27–10.27 emu g−1 depending upon the composition of IL and EG in the binary mixture. The saturation magnetization decrease initially (αFe-0 to αFe-25) followed by an increase (αFe-50–αFe-100) as shown in Fig. 9. It is well known that magnetic properties of ferromagnetic materials are greatly influenced by morphologies and structure of NPs.53 As the size of α-Fe2O3 particles approaches to nanometer scale, due to the nanoscale confinement,54 unusual magnetic behavior has been observed. This behavior is quite different from those of conventional bulk materials (antiferromagnetic at low temperature and ferromagnetic above TM). When particle becomes small enough, the magnetic moment in a single domain fluctuates in a certain direction due to thermal agitation, leading to superparamagnetic behavior above the blocking temperature (TB).55 The increase in the content of IL in the binary mixture leads to decrease in size of NPs. This decrease in size enhances the contribution of uncompensated surface spins and the NPs tend to interact ferromagnetically. Such interactions may lead to enhancement of Ms. The obtained Ms value for the NPs are higher than as reported by Zheng et al.23,26 which can be attributed to small sized NPs resulting in dominance of uncompensated surface spins. Further, the magnetization of smaller NPs randomly flips due to thermal fluctuations which exhibits enhanced magnetization in an external magnetic field and shows the behavior of superparamagnetics.56,57 Thus with increase in the content of IL, the sample αFe-100 shows greater essence of superparamagnetism with enhanced Ms. This observation has also been confirmed from the Mössbauer studies where the spectral area of doublet increases along with increase in BHF for αFe-100. The αFe-0 and αFe-25 NPs also show negligible Mr and Hc like other samples. But the Mössbauer spectra shows only sextet which may be due to shorter time scale for Mössbauer measurements than magnetic measurements. According to Neel–Brown theory, the temperature also affects the superparamagnetic relaxation times.58,59 The super-paramagnetic relaxation time may be in the picoseconds range when the thermal energy is comparable to or larger than the magnetic anisotropy energy. In that case, the main sextet signal is dominant inspite of superparamagnetic signal which might be present in the spectrum. Another possibility for such behavior where superparamagnetic nature has not observed may be based on the particle size distribution in the sample. The smallest particles in the sample have a much lower Debye–Waller factor than the larger particles, resulting in non-observation of superparamagnetic doublet from the smaller particles compared to that of sextet signal from the larger particles in Mössbauer spectrum.60 Some reports are also available in literature where superparamagnetic (doublet) signal is not observed even though the magnetic studies show superparamagnetic nature.61,62 The αFe-0 also shows higher Ms (9.03 emu g−1) which is comparable with αFe-100 (10.27 emu g−1). The possible reasons for such behavior can be the reduction of some Fe3+ to Fe2+ in neat EG, due to weak reducing action of EG, which results in the appearance of ferromagnetic like character due to non-cancellation of spins. Thus superparamagnetic nature of prepared NPs provides a new pathway, particularly in applications where superparamagnetic behavior is advantageous over ferromagnetic behavior.63
 |
| | Fig. 8 (a) Hysteresis loop of α-Fe2O3 NPs synthesized in binary mixture of IL and EG and (b) enlarged view of (a) in the lower range of applied field. | |
 |
| | Fig. 9 Variation of Hc and Ms for the NPs obtained from different compositions of IL and EG. | |
4. Conclusion
The α-Fe2O3 nanoparticles (NPs) have been synthesized by a facile, convenient, effective and fairly low temperature route in the binary mixture of ionic liquid (IL) and ethylene glycol (EG). The characteristic properties of NPs such as crystallinity, size, magnetic and luminescent properties have been found to be dependent on the composition of binary mixture of IL and EG. A decrease in particle size along with decrease in crystallinity of NPs with increase in content of IL has been observed. The α-Fe2O3 NPs prepared in different binary solvent mixtures exhibits weak ferromagnetic character, whereas, the NPs prepared exclusively in binary solvent having higher content of IL (50, 75 and 100%) have shown superparamagnetic character which goes on increasing with increase in the amount of IL. This modulation from weak ferromagnetic to superparamagnetic phase along with luminescent character of the synthesized NPs makes its possible as a good candidate for targeting/delivering specific molecules in living cell under the magnetic field by using a fluorescent microscope in biomedical applications.
Acknowledgements
The authors are very thankful to the Defence Research and Development Organization (DRDO), New Delhi, India for financial support.
References
- N. Pailhé, A. Wattiaux, M. Gaudon and A. Demourgues, J. Solid State Chem., 2008, 181, 2697–2704 CrossRef PubMed.
- L. Huo, W. Li, L. Lu, H. Cui, S. Xi, J. Wang, B. Zhao, Y. C. Shen and Z. Lu, Chem. Mater., 2000, 12, 790–794 CrossRef CAS.
- Z. Y. Fan, X. G. Wen, S. H. Yang and J. G. Lu, Appl. Phys. Lett., 2005, 87, 0131131–0131133 Search PubMed.
- A. Kay, I. Cesar and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- I. Cesar, A. Kay, J. A. G. Martinez and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- T. Ninjbadgar, S. Yamamoto and T. Fukuda, Solid State Sci., 2004, 6, 879–885 CrossRef CAS PubMed.
- A. Watanabe and H. Kozuka, J. Phys. Chem. B, 2003, 107, 12713–12720 CrossRef CAS.
- S. Wagloehner, D. Reichert, D. L. Sorzano, P. Balle, B. Geiger and S. Kureti, J. Catal., 2008, 260, 305–314 CrossRef CAS PubMed.
- A. D. Wheeler, G. Wang, Y. Ling, Y. Li and J. Z. Zhang, Energy Environ. Sci., 2012, 5, 6682–6702 Search PubMed.
- C. Y. Cao, J. Qu, W. S. Yan, J. F. Zhu, Z. Y. Wu and W. G. Song, Langmuir, 2012, 28, 4573–4579 CrossRef CAS PubMed.
- L. Liu, H. Z. Kou, W. Mo, H. Liu and Y. Wang, J. Phys. Chem. B, 2006, 110, 15218–15223 CrossRef CAS PubMed.
- M. Sorescu, R. A. Brand, D. M. Tarabasanu and L. Diamandescu, J. Appl. Phys., 1999, 85, 5546–5548 CrossRef CAS PubMed.
- X. C. Jiang, A. B. Yu, W. R. Yang, Y. Ding, C. X. Xu and S. Lam, J. Nanopart. Res., 2010, 12, 877–893 CrossRef CAS.
- S. B. Wang, Y. L. Min and S. H. Yu, J. Phys. Chem. C, 2007, 111, 3551–3554 CAS.
- D. Dorjnamjin, M. Ariunaa and Y. K. Shim, Int. J. Mol. Sci., 2008, 9, 807–820 CrossRef CAS PubMed.
- J. Dupont, G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner and S. R. Teixeira, J. Am. Chem. Soc., 2002, 124, 4228–4229 CrossRef CAS PubMed.
- D. P. Liu, G. D. Li, Y. Su and J. S. Chen, Angew. Chem., Int. Ed., 2006, 45, 7370–7373 CrossRef CAS PubMed.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS PubMed.
- B. G. Trewyn, C. M. Whitman and V. S. Y. Lin, Nano Lett., 2004, 4, 2139–2143 CrossRef CAS.
- S. W. Cao and Y. J. Zhu, Acta Mater., 2009, 57, 2154–2165 CrossRef CAS PubMed.
- W. J. Zheng, X. D. Liu, Z. Y. Yan and L. J. Zhu, ACS Nano, 2009, 3, 115–122 CrossRef CAS PubMed.
- J. Lian, X. Duan, J. Ma, P. Peng, T. Kim and W. Zheng, ACS Nano, 2009, 3, 3749–3761 CrossRef CAS PubMed.
- C. M. Lee, H. J. Jeong, S. T. Lim, M. H. Sohn and D. W. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 756–759 CAS.
- L. Xu, J. Xia, L. Wang, J. Qian, H. Li, K. Wang, K. Sun and M. He, Chem.–Eur. J., 2014, 20, 2244–2253 CrossRef CAS PubMed.
- J. Ma, T. Wang, X. Duan, J. Lian, Z. Liu and W. Zheng, Nanoscale, 2011, 3, 4372–4375 RSC.
- J. Ma, J. Teo, L. Mei, Z. Zhong, Q. Li, T. Wang, X. Duan, J. Lianc and W. Zheng, J. Mater. Chem., 2012, 22, 11694–11700 RSC.
- L. Xu, J. Xia, K. Wang, L. Wang, H. Li, H. Xu, L. Huangb and M. He, Dalton Trans., 2013, 6468–6477 RSC.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS PubMed.
- A. Mele, C. D. Tran and S. H. D. Lacerda, Angew. Chem., Int. Ed., 2003, 42, 4364–4366 CrossRef CAS PubMed.
- T. Singh, K. S. Rao and A. Kumar, ChemPhysChem, 2011, 12, 836–845 CrossRef CAS PubMed.
- T. Singh, A. Kumar, M. Kaur, G. Kaur and H. Kumar, J. Chem. Thermodyn., 2009, 41, 717–723 CrossRef CAS PubMed.
- W. B. White, J. Ceram. Process Res., 2005, 6, 1–9 Search PubMed.
- K. V. Manukyan, Y. S. Chen, S. Rouvimov, P. Li, X. Li, S. Dong, X. Liu, J. K. Furdyna, A. Orlov, G. H. Bernstein, W. Porod, S. Roslyakov and A. S. Mukasyan, J. Phys. Chem. C, 2014, 118, 16264–16271 CAS.
- H. M. Fan, G. J. You, Y. Li, Z. Zheng, H. R. Tan, Z. X. Shen, S. H. Tang and Y. P. Feng, J. Phys. Chem. C, 2009, 113, 9928–9935 CAS.
- X. D. Liu, J. M. Ma, P. Peng and W. J. Zheng, Mater. Sci. Eng., B, 2008, 150, 89–94 CrossRef CAS PubMed.
- K. Shimizu, C. E. S. Bernardes, A. Triolob and J. N. C. Lopes, Phys. Chem. Chem. Phys., 2013, 15, 16256–16262 RSC.
- Y. P. He, Y. M. Miao, C. R. Li, S. Q. Wang, L. Cao, S. S. Xie, G. Z. Yang, B. S. Zou and C. Burda, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125411–125420 CrossRef.
- J. K. Vassiliou, V. Mehrotra, M. W. Russell, E. P. Giannelis, R. D. McMichael, R. D. Shull and R. F. Ziolo, J. Appl. Phys., 1993, 73, 5109–5116 CrossRef CAS PubMed.
- N. Tsuda, K. Nasu, A. Yanase and K. Siratori, Electronic Conduction in Oxide, ed. M. Cardona, Springer Series in Solid State Science 94, Springer-Verlag, Berlin, 1991 Search PubMed.
- S. H. Tolbert and A. P. Alivisatos, Science, 1994, 265, 373–376 CAS.
- B. Zou, W. Huang, M. Han, S. F. Y. Li, X. Wu, Y. Zhang, J. Zhang, P. Wu and R. Wang, J. Phys. Chem. Solids, 1997, 58, 1315–1320 CrossRef CAS.
- B. Zou, G. Tang, G. Zhang and W. Chen, Acta Phys. Sin., 1993, 42, 1127–1133 CAS.
- P. Ayyub, M. Mullani, M. Barma, R. Palkar and R. Bijayaraghavan, J. Phys. C: Solid State Phys., 1988, 21, 2229–2245 CrossRef CAS.
- L. C. Sanchez, J. D. Arboleda, C. Saragovi, R. D. Zysler and C. A. Barrero, Phys. B, 2007, 389, 145–149 CrossRef CAS PubMed.
- W. Kündig and H. Bömmel, Phys. Rev., 1966, 142, 327–332 CrossRef.
- B. Ganguly, F. E. Huggins, R. P. M. Rao and G. P. Huffman, J. Catal., 1993, 142, 552–560 CrossRef CAS.
- C. Frandsen and S. Mørup, J. Magn. Magn. Mater., 2003, 266, 36–48 CrossRef CAS.
- J. G. Kim, K. H. Han, C. H. Lee and J. Y. Jeong, J. Korean Phys. Soc., 2001, 38, 798–802 CAS.
- S. Mukherjee, D. Das, S. Mukherjee and P. S. Chakrabati, J. Phys. Chem. C, 2010, 114, 14763–14771 CAS.
- X. M. Liu, S. Y. Fu, H. M. Xiao and C. J. Huang, J. Solid State Chem., 2005, 178, 2798 CrossRef CAS PubMed.
- X. Wang, L. Zhang, Y. Ni, J. Hong and X. Cao, J. Phys. Chem. C, 2009, 113, 7003–7008 CAS.
- M. Sorescu, R. A. Brand, D. M. Tarabasanu and L. Diamandescu, J. Appl. Phys., 1999, 85, 5546–5548 CrossRef CAS PubMed.
- H. Arnim, Chem. Rev., 1989, 89, 1861–1871 CrossRef.
- M. Tadić, D. Marković, V. Spasojević, V. Kusigerski, M. Remškar, J. Pirnat and Z. Jagličić, J. Alloys Compd., 2007, 441, 291–296 CrossRef PubMed.
- B. Martínez, X. Obradors, L. Balcells, A. Rouanet and C. Monty, Phys. Rev. Lett., 1998, 80, 181–184 CrossRef.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed. Engl., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- L. Neel, Ann. Geophys., 1949, 5, 99–136 Search PubMed.
- W. F. Brown Jr, Phys. Rev., 1963, 130, 1677–1686 CrossRef.
- R. Alcántara, P. Lavela, G. Ortiz, I. Rodríguez and J. L. Tirado, Hyperfine Interact., 2008, 187, 13–17 CrossRef.
- S. Morup and H. Topsoe, Appl. Phys., 1976, 11, 63–66 Search PubMed.
- M. F. Hansen, F. Bødker, S. Mørup, K. Lefmann, K. N. Clausen and P.-A. Lindgard, Phys. Rev. Lett., 1997, 79, 4910–4913 CrossRef CAS.
- S. K. Suh, K. Yuet, D. K. Hwang, K. W. Bong, P. S. Doyle and T. A. Hatton, J. Am. Chem. Soc., 2012, 134, 7337–7343 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 











