
Acid spike effect in spurs/tracks of the low/high linear energy transfer radiolysis of water: potential implications for radiobiology
Abstract
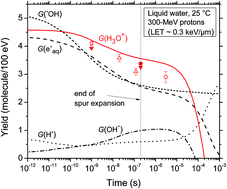
Monte Carlo track chemistry simulations have been used to calculate the yields of hydronium ions (H3O+) that are formed within spurs/tracks of the low/high linear energy transfer (LET) radiolysis of pure, deaerated water during and shortly after irradiation. The in situ formation of H3O+ renders the spur/track regions temporarily more acidic than the surrounding medium. Although experimental evidence for an acidic spur has already been reported, there is only fragmentary information on its magnitude and time dependence. Here, spur/track H3O+ concentrations and the corresponding pH values are obtained from our calculated yields of H3O+ as a function of time (in the interval of ∼1 ps to 1 ms). We selected four impacting ions and we used two different spur/track models: (1) an isolated “spherical” spur model characteristic of low-LET radiation (such as 300 MeV protons, which mimic 60Co γ/fast electron irradiation, LET ∼ 0.3 keV μm−1) and (2) an axially homogeneous “cylindrical” track model for high-LET radiation (such as 150 keV protons, LET ∼ 70 keV μm−1; 1.75 MeV per nucleon helium ions, LET ∼ 70 keV μm−1; and 0.6 MeV per nucleon helium ions, LET ∼ 146 keV μm−1). Very good agreement is found between our calculated time evolution of G(H3O+) in the radiolysis of pure, deaerated water by 300 MeV incident protons and the available experimental data at 25 °C. For all cases studied, an abrupt transient acid pH effect is observed at times immediately after the initial energy release. This effect, which we call an “acid spike”, is found to be greatest for times shorter than ∼1 ns in isolated spurs. In this time range, the pH remains nearly constant at ∼3.3. For cylindrical tracks, the acid spike response to ionizing radiation is far more intense than that for the spherical spur geometry. For the three high-LET irradiating ions considered, the pH is around 0.5 on a time scale of ∼100 ps. At longer times, the pH increases gradually for all cases, ultimately reaching a value of 7 (neutral pH) at ∼1 μs for the spherical geometry and ∼0.1 ms for the cylindrical geometry. It does not appear that the transient acid spike effect described here has been explored in water or in a cellular environment subject to the action of ionizing radiation, especially high-LET radiation. In this regard, this work raises a number of questions about the potential implications of this effect in radiobiology, some of which are briefly evoked.
 Please wait while we load your content...
Please wait while we load your content...

