DOI:
10.1039/C5RA07155K
(Paper)
RSC Adv., 2015,
5, 54920-54928
Influence of the spacer length on the phase behaviors of mesogen-jacketed liquid crystalline polymers with a bulk side-chain†
Received
20th April 2015
, Accepted 16th June 2015
First published on 17th June 2015
Abstract
A series of mesogen-jacketed liquid-crystalline polymers (MJLCPs) containing two triphenylmethyl (Tr) units in the side chains, named poly{2,5-bis[(triphenylmethoxy-alkyl)oxycarbonyl]-styrenes} (denoted as Pv-m-Tr, m = 2, 4, 6, 8, 10, 12, which is the number of the methylene units between the terephthalate core and Tr moieties in the side chains), were designed and successfully synthesized via free radical polymerization. The chemical structures of the monomers were confirmed by 1H NMR and mass spectrometry. The molecular characterizations of the polymers were performed with 1H NMR, GPC and TG analysis. The phase structures and transition temperatures of the polymers were investigated by a combination of techniques including DSC, POM and 1D/2D WAXD. The experimental results revealed that all the polymers exhibited excellent thermal stabilities. The liquid crystalline (LC) phase structures and behaviors of the polymers were strongly dependent on the spacer length (m). With the increase of m, Tg of Pv-m-Tr decreased from 116.7 °C to 11.5 °C because of the alkyl plasticization. When m = 2, 4, 6, 8, 10, the Pv-m-Tr formed a stable columnar nematic (ΦN) phase above Tg. When m = 12, the Pv-12-Tr presented a re-entrant isotropic phase above Tg and a LC phase at higher temperature. This work provides a new case to understand the structure–property relationships of MJLCPs based on the alkyl spacer and bulk side-chain.
1. Introduction
In recent years, a substantial amount of liquid-crystalline polymers (LCPs) have been designed and synthesized because of their potential applications in many fields, including engineering plastics, optical data storage, electro-optics, nonlinear optic devices and so on.1–6 However, most of those applications depend on the phase structures and transitions of LCPs. Therefore, many researchers have investigated the structure–property relationship of LCPs in order to understand the principles of structure formation and structure manipulation.7,8
Generally speaking, LCPs can be divided into main-chain liquid-crystalline polymers (MCLCPs) and side-chain liquid crystalline (SCLCPs) according to the positions of mesogens.9 Depending on the position where the mesogens can be either terminally or laterally attached to the main-chain, the SCLCPs can be categorized into either end-on SCLCPs or side-on SCLCPs. For the end-on SCLCPs, flexible spacers are needed to decouple motions of the main chain and the mesogenic side groups according to Finkelmann's principle.10 However, Zhou et al.11 reported a special type of side-on SCLCPs called mesogen-jacketed liquid crystalline polymers (MJLCPs), which had a short spacer or with a single covalent bond connecting the mesogen to the polymer backbone. The main chain was forced to take an extended chain conformation due to the steric hindrance exerted by the highly populated bulky, rigid side groups, leading to that they display many thermotropic properties characterized by MCLCPs, although MJLCPs belong to SCLCPs chemically, such as high glass transition temperature, broad temperature range of mesophase and forming banded texture after mechanical shearing in LC state.
Phase reentrant behavior is a very unique physical phenomenon in liquid crystal (LC) field. It refers to the phase transition behavior process of the LC material that the same phase structure appears twice or more times under different conditions, which violates the regularity that molecular order should decrease with the increase of temperature.12 In recent years, phase reentrant behaviors have attracted much attention because of their unique potential applications as multifunctional electrical and optical materials.13 So the design and synthesis of materials with the phase reentrant behavior is a hot topic of research. At present, the phase reentrant phenomenon can be observed in the three types LC molecules. First, in the small LC molecules system, it discovered that lauric acid potassium solution showed isotropic reentrant phase during heating in 1980 for the first time by Yu et al.14 Subsequently, some disc and rigid rod-like LC small molecules have appeared isotropic reentrant phase in the process of phase transition.15–18 Second, in the LC oligomer system, Percec et al. reported a re-entrant isotropic phase in a supramolecular disc-like oligomer when the degree of polymerization was four or five.13 Thirdly, in the LC polymers system, the phase reentrant phenomenon was found in the end-on SCLCPs,19–23 polypeptide,24,25 and MJLCPs.26,27
From the previous phase reentrant behaviors of MJLCPs, we were aware of that the tail length greatly influenced on the phase behavior of MJLCPs.26,28–31 For example, our group has synthesized a series of MJLCPs, poly{2,5-bis[(4-alkoxyphenyl)oxycarbonyl]styrenes} (P-OCm, m = 1, 2, 4, 6, 8, 10, 12, 14, 16 and 18) and found that the P-OCm showed the re-entrant isotropic phase when m > 4. Zhu et al.29 and Yu et al.30 have reported the synthesis and characterization of series of MJLCPs, poly[2,5-bis(4′-alkoxycarbonylphenyl)styrene]s and poly[2,5-bis(4′-alkoxyphenyl)-styrene], respectively. When the alkyl tail length exceeded a minimum critical length, both polymers presented the reentrant isotropic phase. However, there were few reports to research the effect of spacer length on phase reentrant behaviors of MJLCPs. At present, only Zhu et al.32,33 has studied the phase behavior of MJLCPs containing two triphenylene (Tp) units in the side chains with the different length of the flexible alkyl spacer (m). The results indicated that the polymers could form the re-entrant isotropic phase when m = 12.
Herein, in order to deep understand the phase reentrant behaviors of MJLCPs, we designed a series of MJLCPs bearing triphenylmethyl group (Tr) with different spacer length, poly{2,5-bis[(triphenylmethoxy-alkyl)oxycarbonyl]-styrenes} (m is the number of the methylene units between the terephthalate core and Tr moieties in the side chains). Compared with the Tp, there was no π–π interaction between Tr and Tr so that the Tr cannot self-organize into well-ordered, self-healable supramolecular structure. However, the Tr contains the bulk space volume due to the three aromatic rings, lead to that the poly(triphenylmethyl methacrylate)[poly(TrMA)] is the optically active vinyl polymer whose chirality arises exclusively from its helical main chain conformation and not from any chiral substituent.34–39 So, when we discussed the formation of LC phase structure of MJLCPs, we only thought the space volume of side-chain. The molecular structure of polymers is shown in Scheme 1. The chemical structures of the monomers were confirmed by 1H NMR and mass spectrometry. The polymers were characterized with 1H NMR, gel permeation chromatography (GPC), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), polarized optical microscopy (POM), and 1D and 2D wide-angle X-ray diffraction (1D and 2D WAXD). To our knowledge, it was first time to synthesize MJLCPs containing triphenylmethyl group with alkyl spacer. Through researching the influence of spacer length on phase behaviors of MJLCPs with bulk side-chain, we obtained some knowledge about the phase reentrant behaviors and the arrangement mode of molecular chain, and then got the essence of the structure of MJLCPs.
 |
| | Scheme 1 Chemical structures of the polymers (Pv-m-Tr). | |
2. Experimental
2.1. Materials
Anhydrous tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl under argon and used immediately. Triethylamine (TEA) and dichloromethane (CH2Cl2) were dried over magnesium sulfate anhydrous. 2,2-Azobisisobutyronitrile (AIBN) was freshly recrystallized from methanol. All other reagents and solvents were used as received without further purification.
2.2. Synthesis of monomers
The chemical structures and synthetic procedures of monomers are illustrated in Scheme 2. For convenience, the monomers of 2,5-bis[(triphenylmethoxy-alkyl)oxycarbonyl]-styrenes were named Mv-m-Tr, where m is the number of the methylene units between the terephthalate core and Tr moieties in the side chains and m = 2, 4, 6, 8, 10, 12 and the corresponding polymers were named Pv-m-Tr. 2-Vinylterephthalic acid (VTA), were facilely synthesized according to the reported procedures.26 The experimental details are described as follows using 2,5-bis[(triphenylmethoxy-hexyl)oxycarbonyl]-styrene (Mv-6-Tr) as an example.
 |
| | Scheme 2 Synthetic route of the monomers and the corresponding polymers. | |
Synthesis of 6-triphenylmethoxy-1-hexanol. According to literature,40 6-triphenylmethoxy-1-hexanol was easy to synthesis and obtain. Hexanediol (12.00 g, 0.1 mol) and CH2Cl2 (100.0 mL) were charged in a 250 mL round-bottomed flask. Then Et3N (4.5 mL, 0.03 mol), DMAP (0.01 g, 0.80 mmol), trityl chloride (6.00 g, 0.02 mol) were added. The mixture was stirred at room temperature for 24 h. Then it was diluted with CH2Cl2 (200.0 mL) and water (100.0 mL). The organic phase was washed with water (100.0 mL) for three times. The organic layer was dried with anhydrous MgSO4, filtered and evaporated. The purified product was obtained by passing through a silica gel column with ethyl acetate and petroleum ether (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as eluent. The final product of 6-triphenylmethoxy-1-hexanol was white solid. Yield: 71%. 1H NMR (δ, ppm, CDC13): δ = 1.10–1.47 (m, 4, –CH2–, and 1H, –OH), 1.48–1.71 (m, 4H, –CH2–), 2.95–3.12 (m, 2H, –OCH2–), 3.55–3.72 (m, 2H, –OCH2–), 7.15–7.55 (m, 15H, Ar-H).
:3) as eluent. The final product of 6-triphenylmethoxy-1-hexanol was white solid. Yield: 71%. 1H NMR (δ, ppm, CDC13): δ = 1.10–1.47 (m, 4, –CH2–, and 1H, –OH), 1.48–1.71 (m, 4H, –CH2–), 2.95–3.12 (m, 2H, –OCH2–), 3.55–3.72 (m, 2H, –OCH2–), 7.15–7.55 (m, 15H, Ar-H).
Synthesis of 2,5-bis[(triphenylmethoxy-hexyl)oxycarbonyl]-styrene (Mv-6-Tr). The experimental details were described as follows: 6-triphenylmethoxy-1-hexanol (3.00 g, 8.40 mmol), VTA (0.80 g, 4.20 mmol), N,N′-dicyclohexylcarbodiimide (DCC, 3.40 g, 16.80 mmol), 4-(dimethylamino)pyridine (DMAP, 5.0 mg, 0.04 mmol), and dried CH2Cl2 (100.0 mL) were mixed in a 250 mL round-bottomed flask and stirred at ambient temperature for 24 h. The floating solid was filtrated and the solvent was evaporated under reduced pressure. The crude product was purified by silica gel column chromatography with ethyl acetate and petroleum ether (v/v, 1:5) as eluent, and then condensed eluent to yield a colorless liquid, yield: 55%. 1H NMR (δ, ppm, CDC13): δ = 1.32–1.51 (d, 8H, –CH2–), 1.59–1.70 (m, 4, –CH2–), 1.70–1.85 (m, 4H, –CH2–), 3.00–3.12 (t, 4H, –OCH2–), 4.24–4.40 (m, 4H, –OCH2–), 5.35-5.45 (d, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.67–5.80 (d, 1H, CH2), 7.15–7.35 (m, 30H, Ar-H and 1H, –CH), 7.82–7.96 (m, 2H, Ar-H), 8.18–8.26 (s, 1H, Ar-H). Mass Spectrometry (MS) (m/z) [M + Na]+ calcd for C60H60O6Na, 899.429.; found, 899.711.
CH2), 5.67–5.80 (d, 1H, CH2), 7.15–7.35 (m, 30H, Ar-H and 1H, –CH), 7.82–7.96 (m, 2H, Ar-H), 8.18–8.26 (s, 1H, Ar-H). Mass Spectrometry (MS) (m/z) [M + Na]+ calcd for C60H60O6Na, 899.429.; found, 899.711.
Synthesis of 2,5-bis[(triphenylmethoxy-alkyl)oxycarbonyl]-styrenes (Mv-m-Tr, m = 2, 4, 8, 10, 12). All the other monomers were synthesized and characterized in a similar way. The characterization data of Mv-m-Tr (m = 2, 4, 8, 10, 12) were shown in ESI.†
2.3. Synthesis of polymers
All polymers (Pv-m-Tr in Scheme 2) were obtained by conventional solution free radical polymerization. A typical polymerization procedure was carried out as the following using Pv-6-Tr as an example. A total of 300 mg (0.34 mmol) of Mv-6-Tr, 56 μL of THF solution of 10 mg mL−1 AIBN, 0.70 g of THF, and a magnetic stir bar were added into a polymerization tube. After three freeze–pump–thaw cycles, the tube was sealed off under vacuum. Polymerization was carried out at 70 °C for 10 h. The tube was then opened, and the reaction mixture was diluted with 8.0 mL of THF. Then, the resultant polymer was dropped slowly into the mixture of methanol/THF (3/1). The dissolution and precipitation were repeated three times. After drying under vacuum, 0.18 g of polymer was obtained. Yield: 60%.
2.4. Instruments and measurements
Nuclear magnetic resonance (NMR). 1H NMR measurements were performed on a Bruker ARX400 MHz spectrometer using with CDCl3 as solvent, tetramethylsilane (TMS) as the internal standard at room temperature.
Gel permeation chromatography (GPC). The apparent number average molecular weight (Mn) and polydispersityindex (PDI = Mw/Mn) were measured on a GPC (WATERS 1515) instrument with a set of HT3, HT4 and HT5. The μ-styragel columns used THF as an eluent and the flow rate was 1.0 mL min−1 at 38 °C. The GPC data were calibrated with polystyrene standards.
Thermogravimetric analysis (TGA). TGA was performed on a TA SDT 2960 instrument at a heating rate of 20 °C min−1 in nitrogen atmosphere.
Differential scanning calorimetry (DSC). DSC traces of the polymer were obtained using a TA Q10 DSC instrument. The temperature and heat flow were calibrated using standard materials (indium and zinc) at a cooling and heating rates of 10 °C min−1. The sample with a typical mass of about 5 mg was encapsulated in sealed aluminum pans.
Polarizing optical microscope (POM). LC texture of the polymer was examined under POM (Leica DM-LM-P) equipped with a Mettler Toledo hot stage (FP82HT).
One-dimensional wide-angle X-ray diffraction (1D WAXD). 1D WAXD experiments were performed on a BRUKER AXS D8 Advance diffractometer with a 40 kV FL tubes as the X-ray source (Cu Kα) and the LYNXEYE_XE detector. Background scattering was recorded and subtracted from the sample patterns. The heating and cooling rates in the 1D WAXD experiments were 10 °C min−1.
Two-dimensional wide-angle X-ray diffraction (2D WAXD). 2D WAXD was carried out using a BRUKER AXS D8 discover diffractometer with a 40 kV FL tubes as the X-ray source (Cu Kα) and the VANTEC 500 detector. The point-focused X-ray beam was aligned either perpendicular or parallel to the mechanical shearing direction. For both the 1D and 2D WAXD experiments, the background scattering was recorded and subtracted from the sample patterns.
3. Results and discussion
3.1. Synthesis and characterization of monomers and polymers
As shown in Scheme 2, the monomers were synthesized in only two steps. The structures of the monomers were confirmed by conventional analyses, including 1H NMR and mass spectrometry.
All the monomers could be easily polymerized via free radical polymerization. Herein, we use Pv-6-Tr as an example to elucidate the process. Fig. 1(a) and (b) give the 1H NMR spectra (CDCl3−d) of the monomer Mv-6-Tr and the polymer Pv-6-Tr, respectively. The Mv-6-Tr showed the characteristic resonances of the vinyl group at 5.35–5.80 ppm. After polymerization, these signals disappeared completely. The chemical shifts of Pv-6-Tr were quite broad and consistent with the expected polymer structure. The apparent number-average molecular weights of the polymers determined by GPC were higher than 5 × 104 g mol−1, demonstrating good polymerizability of the monomers. The molecular characterizations of the polymers are summarized in Table 1.
 |
| | Fig. 1 1H NMR spectrums of the monomer Mv-6-Tr (a) and the polymer Pv-6-Tr (b) in CDCl3. | |
Table 1 Molecular characteristics and properties of the series of Pv-m-Tr
| Sample |
Mn (×10−4)a |
DPa |
PDIa |
Tdb (°C) |
Tgc (°C) |
T1d (°C) |
| The apparent number-average molecular weight (Mn), average degree of polymerization (DP) and polydispersityindex (PDI) were measured by GPC using PS standards. The temperatures of 5% weight loss under nitrogen were measured by TGA heating experiments at a rate of 20 °C min−1. The glass transition temperatures were measured by DSC at a heating rate of 10 °C min−1 under a nitrogen atmosphere during the second heating process. The transition temperature from isotropic phase to the liquid crystalline phase measured by POM at a heating and cooling rate of 2 °C min−1. |
| Pv-2-Tr |
6.73 |
88 |
1.80 |
359 |
116.7 |
|
| Pv-4-Tr |
5.40 |
66 |
2.20 |
362 |
69.7 |
|
| Pv-6-Tr |
6.70 |
76 |
1.87 |
362 |
47.9 |
|
| Pv-8-Tr |
5.56 |
60 |
2.09 |
365 |
31.7 |
|
| Pv-10-Tr |
16.43 |
166 |
2.43 |
370 |
20.3 |
|
| Pv-12-Tr |
15.28 |
146 |
2.30 |
374 |
11.5 |
260 |
3.2. Phase transitions and phase structures of the monomers and polymers
Among all the monomers synthesized, Mv-m-Tr (m = 4, 6, 8, 10, 12) are liquid at the room temperature. Mv-2-Tr is crystalline with a melting temperature of 135 °C.
The thermal and liquid-crystalline properties of the polymers were investigated by TGA, DSC, and POM. As shown in Table 1, all polymers exhibited excellent thermal stabilities with the temperatures at 5% weight loss about 360 °C in nitrogen.
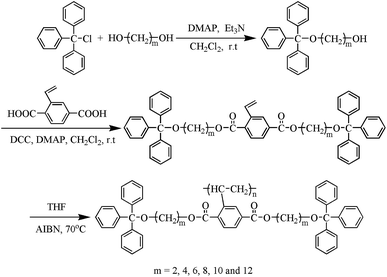
The phase-transition behaviors of the polymers were investigated by DSC. The samples firstly were heated from ambient temperature to 300 °C at a rate of 20 °C min−1 under nitrogen atmosphere to eliminate the thermal history. Fig. 2 shows the first cooling and the second heating DSC curves of Pv-m-Tr (m = 2, 4, 6, 8, 10, 12) at a rate of 10 °C min−1. All samples exhibited a single and obvious glass transition (Tg) when heating or cooling. As expected, Tg decreases from 116.7 °C to 11.5 °C as the spacer length increases owing to the strong internal plasticization effect of the longer alkyl spacers and the results are listed in Table 1. Except Tg, no other transition peak was observed above Tg or below Tg (see Fig. S1†) and this phenomenon was often found in the other MJLCPs.41–43
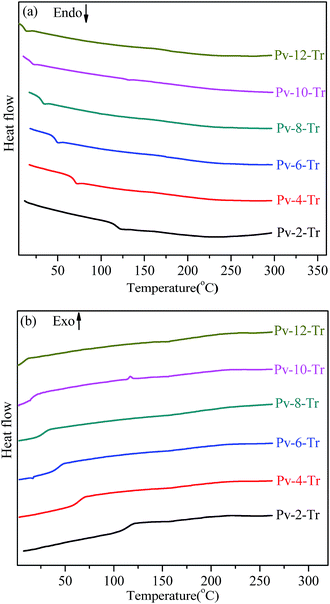 |
| | Fig. 2 DSC curves of Pv-m-Tr during the second heating scan (a) and the first cooling scan (b) at a rate of 10 °C min−1 under nitrogen atmosphere. | |
Birefringence of the polymers was observed by POM. All samples were cast from THF solution and slowly dried at room temperature, and then slowly heated. The results showed that the polymers can be divided into two types. The first one is the Pv-m-Tr (m = 2, 4, 6, 8 and 10). They exhibited strong birefringence that always remained even when heated to 300 °C. When cooled to room temperature from 300 °C, the birefringence of the sample also remained unchanged. All these polymers showed the stable needle texture and the POM image of polymer Pv-6-Tr is shown in Fig. 3(a), implying the formation of columnar phase. Second one is Pv-12-Tr. On heating, the sample became soft above the Tg, but there was no birefringence to be seen [Fig. 3(b)], indicating isotropic phase formed. Of course, there is the possibility that the optically isotropic phase is not a true re-entrant isotropic liquid phase, but some kind of cubic phase analogous to the smectic D phase in rod-like liquid crystals. However, no birefringence could be detected by rotating the sample or by inducing shear flow by moving the upper glass slide. These observation suggest that we are dealing with a normal isotropic liquid phase and make it less likely that it is some kind of cubic phase. Moreover, XRD further proved that the polymer formed the isotropic phase. When the temperature reached about 260 °C, the needle texture was observed [Fig. 3(c)], suggesting the polymers presented the columnar phase. On heating further, no visible change of birefringence was observed before decomposition (onset temperature > 300 °C). On the subsequent cooling, the original scene was reestablished, that was, the birefringence disappeared. Further cooling to −10 °C which was below Tg, no birefringence was still observed. Therefore, based on our previous research,26 the former polymers belonged to the transitional MJLCPs. The later polymers were the first kind of MJLCPs with a re-entrant phase.
 |
| | Fig. 3 Representative POM images of the texture of the Pv-6-Tr maintained at 200 °C (a), and representative POM images of the texture of the Pv-12-Tr maintained at 200 °C (b) and 260 °C (c). | |
To get more insight into the phase transition temperature, we performed reflection light intensity (birefringence) measurements. Although the phase transition process from isotropic phase to LC phase could not be detected by DSC because of the thermal heat was small, it could detected by reflection light intensity measurements. Fig. 4 shows the recorded changes in reflection light intensity for Pv-6-Tr and Pv-12-Tr by POM at a rate of 10 °C min−1. As can been seen, there is little change for the intensity of Pv-6-Tr and the reflection light intensity kept at 65%, indicating no change of phase structure below 300 °C. However, an obvious transition peak appeared at 260 °C (T1) in which the reflection light intensity sharply increased to 68% from 12%, showing a transition from isotropic state to ordered state.44
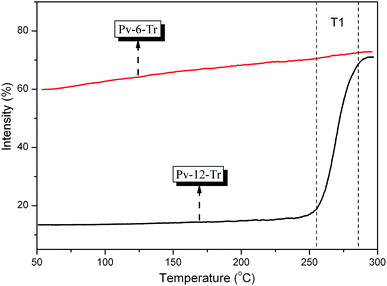 |
| | Fig. 4 The change of reflection light intensity for Pv-6-Tr and Pv-12-Tr at a heating rate 10 °C min−1 in POM. | |
3.3. Phase structure identification of the polymer
In order to study the mesomorphic structure of the polymers, temperature dependant 1D WAXD experiment was conducted. A sample for 1D WAXD experiments was about 60 mg of the polymer was added into an aluminum foil substrate. In order to be consistent with the DSC and POM results, the samples were heated to 300 °C, and then slowly cooled to the room temperature. The temperature was not higher than 300 °C during the experiments process for the sample to avoid thermal degradation. Fig. 5(a) and (b) illustrates the temperature-variable 1D WAXD patterns of Pv-2-Tr from 30 to 210 °C and from 210 to 30 °C during the second heating and subsequent cooling. And the test region of 2θ was from 1.5° to 35°. At low angle (2θ < 10°), one narrow reflection peak was observed at 30 °C, indicating the ordered structures on the nanometer scale formed (2θ = 4.29°, which a d-spacing value of 2.06 nm). The intensity of the halo basically kept the same with the temperature elevated, i.e., 210 °C, which was higher than the Tg of the polymer, only the peak shifted slightly to high 2θ value due to the thermal expansion. When cooling, the reflection peak intensity unchanged, suggesting that polymer always kept the ordered structures whatever heating or cooling. No higher order diffractions of the low-angle peak can be observed. In the wide-angle region of 2θ > 10°, two amorphous halos located in the 2θ values of ∼12° (d ≈ 0.74 nm) and ∼20° (d ≈ 0.44 nm) whether in the heating or cooling process, which reflected that no long range ordered structure formed via molecular packing was detected over the entire temperature region studied. As the temperature increased, the two peaks intensity gradually decreased and the centre of the two halos slightly shifted to lower 2θ angles caused by the thermal expansion. Due to the lack of higher-order diffractions in the low angle or high angle, the polymer exhibited a low-ordered LC phase. The structural characteristics (the jacketed structure and the simulated length of the bulky side group, 2.57 nm) of the polymer and the needle-like texture indicated that the LC phase was probably a ΦN developed by the main chain and Tr as a whole. Similar to the cases of typical MJLCPs, the low angle peak corresponded to the columnar diameter (2.06 nm), which was determined mainly by the length of the side-chain mesogen.27,45 It is noted that Pv-2-Tr rod diameter of 2.06 nm in the columnar (ΦN) phase was less than the calculated length of the side group (2.57 nm). Therefore, the side chains tilt around 37°. This angle is also agreement with other MJLCPs.46 For the scattering halo at 2θ = 20° (d spacing of 0.44 nm), it should correspond to the spacing between the side groups, but for the scattering halo at 2θ = 12°, we still did not get a reasonable interpretation about the packing, which may be caused by Tr.
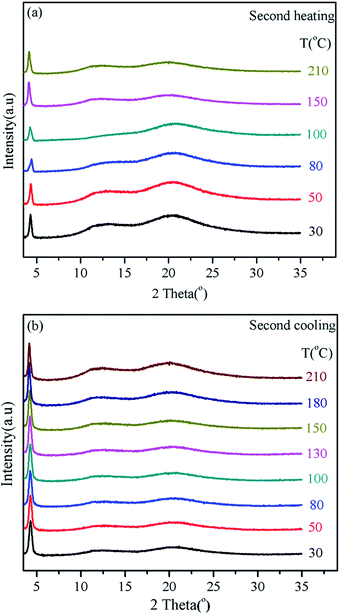 |
| | Fig. 5 1D WAXD patterns of Pv-2-Tr during the second heating (a) and subsequent cooling (b). | |
In addition, the polymers Pv-m-Tr (m = 4, 6, 8 and 10) showed similar results with Pv-2-Tr in 1D WAXD experiments, which was consistent with the POM result. For the four samples, the changes and positions of the two scattering halos in the high 2θ region of 10–30° were similar. However, the d-spacing of the polymers in the low 2θ region slightly increased as the length of alkyl spacer increased, for example, at 30 °C, the d spacing of the polymer Pv-4-Tr was 2.18 nm (2θ = 4.05), but for polymer Pv-6-Tr was 2.33 nm (2θ = 3.80), polymer Pv-8-Tr was 2.54 nm (2θ = 3.48), and for polymer Pv-10-Tr was 2.62 nm (2θ = 3.37). 1D WAXD patterns of all the Pv-m-Tr (m = 2, 4, 6, 8 and 10) at 30 °C are presented in Fig. 6 and the corresponding d-spacing values of the low-angle peak/halo are summarized in Table 2.
 |
| | Fig. 6 1D WAXD patterns of Pv-m-Tr (m = 2, 4, 6, 8 and 10) at 30 °C and Pv-12-Tr at 280 °C. | |
Table 2 The 2θ d-spacing values and calculated length of the side-chain
| Sample |
2θa (o) |
d-spacinga (nm) |
Calculated length of the side-chainb (nm) |
| Obtained from the one-dimensional WAXD experiments. Assuming the n-alkyl spacer in the side chains have an all-trans conformation. |
| Pv-2-Tr |
4.29 |
2.06 |
2.57 |
| Pv-4-Tr |
4.05 |
2.18 |
3.13 |
| Pv-6-Tr |
3.80 |
2.33 |
3.62 |
| Pv-8-Tr |
3.48 |
2.54 |
4.11 |
| Pv-10-Tr |
3.37 |
2.62 |
4.53 |
| Pv-12-Tr |
3.13 |
2.82 |
5.15 |
Compared with 1D WAXD patterns of Pv-m-Tr (m = 2, 4, 6, 8 and 10), the Pv-12-Tr showed the different 1D WAXD patterns. Below 240 °C, the low-angle diffraction peak is diffuse and weak [see Fig. 7(a)]. At temperatures above 240 °C, a sharp and intense peak at 2θ = 3.13° (d spacing of 2.82 nm) appears. With the further increasing temperature, the intensity of the peak increases. This peak becomes weak and disappears at 240 °C on the following cooling scan [see Fig. 7(b)].The distinct discontinuous intensities correspond with the endothermal transition of POM at about 260 °C during the second heating. The discontinuous observation during cooling procedure clearly indicated that the LC phase formed at higher temperature disappeared during cooling. Those implied that the Pv-12-Tr formed the re-entrant phase.47 This experiment results were observed in some MJLCPs, such as, poly[di(alkyl)vinylterephthalates], poly{2,5-bis[(4-alkoxyphenyl)oxycarbonyl]styrenes}, poly[2,5-bis(4′-alkoxycarbonylphenyl)styrene]s and poly[2,5-bis(4′-alkoxyphenyl)-styrene], poly{2,2,3,3,4,4,4-heptafluorobutyl 4′-hydroxy-2-vinylbiphenyl-4-carboxylate}, poly{butyl 4′-hydroxy-2-vinylbiphenyl-4-carboxylate}, poly(alkyl 4′-(octyloxy)-2-vinylbiphenyl-4-carboxylate) and so on.26–28,45
 |
| | Fig. 7 1D WAXD patterns of Pv-12-Tr during the second heating (a) and subsequent cooling (b). | |
To further confirm the phase structures of the polymers, 2D WAXD experiments were carry out. All fiber samples were drawn with a pair of tweezers at above glass-transition temperature. Fig. 8 shows the 2D WAXD patterns of the polymer Pv-2-Tr at room temperature with X-ray incident beam perpendicular and parallel to the fiber direction. In Fig. 8(a) the X-ray beam was perpendicular to the shear direction that was along the meridian, a pair of strong diffraction arcs was observed on the equator at 2θ = 4.29° (d spacing was 2.06 nm), indicating that the order structure was developed perpendicular to the fiber axis on the nanometer scale. This was perfectly consistent with the 1D-WAXD results. When the X-ray incident beam was parallel to the shear direction, a ring pattern at 2θ = 4.36° (d spacing of 2.04 nm) was present, which exhibited an isotropic intensity distribution [Fig. 8(b)]. Meanwhile, two diffusion scattering halos in the high 2θ angle were observed, suggesting only the short-range order exists. Considering the similar X-ray results reported previously, we proposed the polymer Pv-2-Tr exhibited a columnar nematic phase.27,45 The polymers Pv-m-Tr (m = 4, 6, 8 and 10) showed similar 2D WAXD experiment results, which were considered to generate columnar nematic phase, in which each cylinder is attributed to a single polymer chain with the side groups tightly jacketing the backbone (see Fig. 9). For the Pv-12-Tr, we couldn't obtain 2D WAXD patterns even though the sample heated to 280 °C, then quickly put into the liquid nitrogen to want to keep the LC phase structure.
 |
| | Fig. 8 2D WAXD fiber patterns of Pv-2-Tr. The X-ray incident beam was perpendicular (a) and parallel (b) to the fiber axis. | |
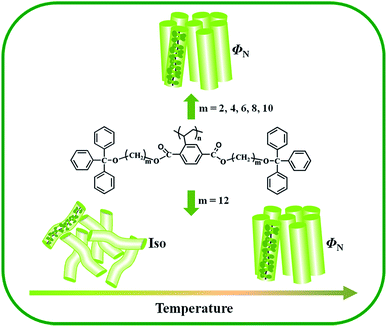 |
| | Fig. 9 Schematic drawing of the thermotropic phase behavior of the polymers. | |
3.4. Influence of the spacer length on the phase behaviors
Based on the results of DSC, POM, 1D WAXD and 2D WAXD experiments, Pv-m-Tr (m = 2, 4, 6, 8, 10 and 12) exhibits two types of different behaviors with the increasing length of alkyl spacer, as shown in Fig. 9. The one is that polymers Pv-m-Tr (m = 2, 4, 6, 8, 10) show a stable ΦN phase whatever heating or cooling. The other one is that the Pv-12-Tr presents a re-entrant isotropic phase in the low temperature and a ΦN phase in the high temperature. According to the literature, when m = 3, 4, the poly[di(alkyl)vinylterephthalates] (P-m) [see Fig. 10(a)] formed the stable ΦH phase.31 When m = 6, 8, 10, the P-m exhibited the re-entrant isotropic phase. When m = 12, the polymer showed amorphous structure. For the poly{2,5-bis[(3,6,7,10,11-pentakis(hexyloxy)triphenylen)-2-oxyalkyloxycarbonyl]styrene} (PPmV) [see Fig. 10(b)],32,33 the polymers produced rectangular columnar (ΦR) phases at high temperatures. However, at low temperatures, Tp moieties in the side chains formed a discotic nematic phase in conjunction with the ΦR phase developed by the rod-like supramolecular mesogen – the MJLCP chain as a whole, owing to the self-organization of the Tp moieties. When m = 12, a re-entrant isotropic phase is found in the medium temperature range. Therefore, compared with the phase behavior and structure of three kinds of polymers, we understood that, when the side chain contained the bulk group (Tp or Tr), the polymers still formed the stable columnar phase although the alkyl spacer length was long. As for this, the strong steric hindrance was imposed by the highly crowded bulky side groups, lead to force the main-chain to be well extended. Secondly, further increasing the alkyl spacer length, the polymers showed the isotropic phase, which could be described as a random coil because that the steric effect of side-chain became weak in the low temperature. As temperature increased, the backbone adopt a somewhat extended conformation due to the consecutive tempestuously motions of side-chain around the main-chain,48 lead to produce the strong dynamic steric effect. Lastly, the Pv-m-Tr only formed the ΦN phase own to lack the alkyl tail of Tr, which couldn't make the column closely packed.
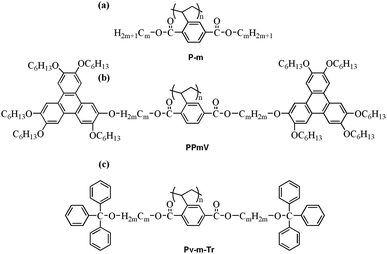 |
| | Fig. 10 Chemical structure of P-m (a), PPmV (b) and Pv-m-Tr (c). | |
4. Conclusions
We have successfully synthesized a series of vinyl monomers Mv-m-Tr (m = 2, 4, 6, 8, 10, and 12) containing different spacer length between the terephthalate core and Tr moieties in the side chains. Upon using conventional radical polymerization, we further obtained the corresponding polymers (Pv-m-Tr). The chemical structures of the monomers and polymers were confirmed by various characterization techniques. The phase structures and transitions of these polymers were investigated by the combined technology of DSC, POM, and XRD. The results of the experiments indicated that the length of alkyl spacer played an important role in the phase behaviors of these polymers with bulk side chain. With the increasing of m, the Tg of Pv-m-Tr (m = 2, 4, 6, 8, 10, and 12) decreased from 116.7 °C to 11.5 °C and the cylinder diameter of Pv-m-Tr increased from 2.06 nm to 2.82 nm. A re-entrant isotropic phase was found when m reached to 12. The phase behavior and the columnar phase structure size were readily tuned by the variation of alkyl spacer, which provided a facile way to tailor the structure and properties of the polymers in real application.
Acknowledgements
This research was financially supported by the National Nature Science Foundation of China (51373148).
References
- L. C. Gao, Z. H. Shen, X. H. Fan and Q. F. Zhou, Polym. Chem., 2012, 3, 1947–1957 RSC.
- X. F. Chen, Z. H. Shen, X. H. Wan, X. H. Fan, E. Q. Chen, Y. G. Ma and Q. F. Zhou, Chem. Soc. Rev., 2010, 39, 3072–3101 RSC.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4176 CrossRef CAS PubMed.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H.-W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 419, 384–387 CrossRef CAS PubMed.
- B. Ni, J. Q. Liao, S. Chen and H. L. Zhang, RSC Adv., 2015, 5, 9035–9043 RSC.
- J. F. Ban, S. Chen and H. L. Zhang, RSC Adv., 2014, 4, 54158–54167 RSC.
- Q. F. Zhou, H. M. Li and X. D. Feng, Mol. Cryst. Liq. Cryst., 1988, 155, 73–82 CrossRef CAS PubMed.
- H. Finkelmann, H. Ringsdorf and J. H. Wendorff, Makromol. Chem., 1978, 179, 273 CrossRef CAS PubMed.
- Q. F. Zhou, H. M. Li and X. D. Feng, Macromolecules, 1987, 20, 233–234 CrossRef CAS.
- N. H. Tinh, J. Malthete and C. Destrade, J. Phys., Lett., 1981, 42, 417–419 CrossRef.
- V. Percec, M. Lee, J. Heck, H. E. Blackwell, G. Ungar and A. Alvarez-Castillo, J. Mater. Chem., 1992, 2, 931–938 RSC.
- L. J. Yu and A. Saupe, Phys. Rev. Lett., 1980, 45, 1000–1003 CrossRef CAS.
- C. Destrade, P. Foucher, J. Malthete and N. H. Tinh, Phys. Lett. A, 1982, 88, 187–190 CrossRef.
- T. W. Warmerdam, R. J. M. Nolte, D. Frenkel and R. J. J. Zijlstra, Liq. Cryst., 1988, 3, 1087–1104 CrossRef CAS PubMed.
- A. Glebowska, P. Przybylski, M. Winek, P. Krzyczkowska, A. Krowczynski, J. Szydlowska, D. Pociecha and E. Gorecka, J. Mater. Chem., 2009, 19, 1395–1398 RSC.
- W. Weissflog, G. Pelzl, I. Letko and S. Diele, Mol. Cryst. Liq. Cryst., 1995, 260, 157–170 CrossRef CAS PubMed.
- P. L. Barny, J. Dubois, C. C. Frledrich and C. Noël, Polym. Bull., 1986, 15, 341–348 CrossRef.
- N. Lacoudre, A. L. Borgne, N. Spassky, J. P. Vairon and C. Noël, Mol. Cryst. Liq. Cryst., 1988, 155, 113–127 CrossRef CAS PubMed.
- V. Percec and D. Tomazos, Adv. Mater., 1992, 4, 548–561 CrossRef CAS PubMed.
- N. Tomikawa, I. Itoh and J. Watanabe, Jpn. J. Appl. Phys., 2005, 44, 381–384 CrossRef.
- B. I. Ostrovskii, S. N. Sulyanov, N. I. Boiko, V. P. Shibaev and W. H. Jeu1, Eur. Phys. J. E, 2001, 6, 277–285 CrossRef CAS PubMed.
- J. P. Lin, Polymer, 1997, 38, 4837–4841 CrossRef CAS.
- R. Subramanian and D. B. Dupre, J. Chem. Phys., 1984, 81, 4626–4629 CrossRef CAS PubMed.
- S. Chen, C. K. Jie, H. L. Xie and H. L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3923–3935 CrossRef CAS PubMed.
- G. L. Jiang, H. H. Cai, Z. H. Shen, X. H. Fan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 557–564 CrossRef CAS PubMed.
- Y. F. Zhao, X. H. Fan, X. H. Wan, X. F. Chen, Y. Yi, L. S. Wang, X. Dong and Q. F. Zhou, Macromolecules, 2006, 39, 948–956 CrossRef CAS.
- Z. G. Zhu, J. G. Zhi, A. H. Liu, J. X. Cui, X. H. Wan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 830–847 CrossRef CAS PubMed.
- Z. N. Yu, H. L. Tu, X. H. Wan, X. F. Chen and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1454–1464 CrossRef CAS PubMed.
- X. Y. Yin, C. Ye, X. Ma, E. Q. Chen, X. Y. Qi, X. F. Duan, X. H. Wan, S. Z. Cheng and Q. F. Zhou, J. Am. Chem. Soc., 2003, 125, 6854–6855 CrossRef CAS PubMed.
- Y. F. Zhu, H. J. Tian, H. W. Wu, D. Z. Hao, Y. Zhou, Z. H. Shen, D. C. Zou, P. C. Sun, X. H. Fan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 295–304 CrossRef CAS PubMed.
- Y. F. Zhu, X. L. Guan, Z. H. Shen, X. H. Fan and Q. F. Zhou, Macromolecules, 2012, 45, 3346–3355 CrossRef CAS.
- T. Nakano, Y. Okamoto and K. Hatada, J. Am. Chem. Soc., 1992, 114, 1318–1329 CrossRef CAS.
- N. Hoshikawa, Y. Hotta and Y. Okamoto, J. Am. Chem. Soc., 2003, 125, 12380–12381 CrossRef PubMed.
- C. Merten and A. Hartwig, Macromolecules, 2010, 43, 8373–8378 CrossRef CAS.
- Y. Okamoto, K. Suzuki and K. Ohta, J. Am. Chem. Soc., 1979, 101, 4763–4765 CrossRef CAS.
- Y. Okamoto, S. Honda, E. Yashima and H. Yuki, Chem. Lett., 1983, 12, 1221–1224 CrossRef.
- T. Sanji, K. Takase and H. Sakurai, J. Am. Chem. Soc., 2001, 123, 12690–12691 CrossRef CAS.
- A. I. Hernández, J. Balzarini, A. Karlsson, M. J. Camarasa and M. J. Pérez-Pérez, J. Med. Chem., 2002, 45, 4254–4263 CrossRef PubMed.
- Q. K. Zhang, H. J. Tian, C. F. Li, Y. F. Zhu, Y. Liang, Z. Shen and X. H. Fan, Polym. Chem., 2014, 5, 4526–4533 RSC.
- L. Weng, J. J. Yan, H. L. Xie, G. Q. Zhong, S. Q. Zhu, H. L. Zhang and E. Q. Chen, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1912–1923 CrossRef CAS PubMed.
- X. Mei, J. Zhang, Z. H. Shen and X. H. Fan, Polym. Chem., 2012, 3, 2857–2866 RSC.
- S. Chen, X. Shu, H. L. Xie and H. L. Zhang, Polymer, 2013, 54, 3556–3565 CrossRef CAS PubMed.
- L. Y. Zhang, H. L. Wu, Z. H. Shen, X. H. Fan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3207–3217 CrossRef CAS PubMed.
- C. Ye, H. L. Zhang, Y. Huang, E. Q. Chen, Y. L. Lu, D. Y. Shen, X. H. Wan, Z. H. Shen, S. Z. D. Cheng and Q. F. Zhou, Macromolecules, 2004, 37, 7188–7196 CrossRef CAS.
- S. Chen, H. B. Luo, H. L. Xie and H. L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 924–935 CrossRef CAS PubMed.
- S. T. Sun, H. Tang, P. Y. Wu and X. H. Wan, Phys. Chem. Chem. Phys., 2009, 11, 9861–9870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07155k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 











