
The mechanism of 2,4,6-trinitrotoluene detection with amino acid-capped quantum dots: a density functional theory study†
Zhaoyang Loua,
Yingqi Cuia,
Mingli Yang*a and
Jun Chen*b
aInstitute of Atomic and Molecular Physics, Key Laboratory of High Energy Density Physics and Technology of Ministry of Education, Sichuan University, Chengdu 610065, China. E-mail: myang@scu.edu.cn; jun_chen@iapcm.ac.cn
bBeijing Institute of Applied Physics and Computational Mathematics, Beijing 100081, China
First published on 15th May 2015
Abstract
When an amino acid-capped quantum dots solution meets 2,4,6-trinitrotoluene (TNT), it changes from colorless to red and its fluorescence is quenched. This is a recently developed technique for the detection of TNT in trace amounts. However, what causes the changes in coloration and fluorescence remains controversial. Using density functional theory calculations, we studied the structures and optical properties of the products of TNT reacting with cysteine. Two compounds, namely, a Meisenheimer complex and a TNT anion, which are obtained from an addition reaction and an acid–base reaction respectively, were characterized, but neither of them could be used solely to interpret the experimental results. Our calculations proposed the possibility of their coexistence in the solution from their similar thermodynamic stability and their predicted absorption and vibrational spectra. The superposition of their calculated optical absorption spectra produces band distributions similar to those of the experiments. Moreover, the measured Raman spectra that had been used to characterize the formation of the Meisenheimer complex cannot exclude the formation of the TNT anion whose characteristic vibrations are buried by those of the former. Our calculations also revealed that in the Meisenheimer complex the electron delocalization in the phenyl ring of TNT is blocked by the attached cysteine, while for the TNT anion, the removal of the hydrogen atom enhances the electron delocalization and leads to a redshift of its first excitation in comparison with that of the Meisenheimer complex. Therefore, the key to TNT detection is to control the size of the QDs so as to adjust their emission to a wavelength around the absorption bands of either the Meisenheimer complex or the TNT anion that are formed with TNT and the amino acid.
1 Introduction
2,4,6-Trinitrotoluene (TNT) is an important energetic material widely used in industrial, agricultural and military affairs. It has definitely affected our lives, from social and personal security to environmental ecology.1–3 Its residues have been detected in soil, groundwater, and even in various food chains, threatening human health because of their high toxicity and denaturation to our bodies.4–6 A simple, sensitive, and cheap method to detect TNT is therefore desired.To date, TNT detection is mainly based on fluorescence,7–21 Raman scattering22–29 and electrochemical30–36 techniques and among these the fluorescence detection technique has attracted considerable attention recently owing to its strengths of high signal output, sensitivity and simplicity.37–40 The rationale of fluorescence detection is that the fluorescence of a reagent changes, shifts or is quenched when it meets TNT. TNT is electron-deficient due to the presence of three nitro groups and tends to interact with electron-rich materials such as some amino acids through a charge-transfer interaction or reaction.41 Many experimental works have reported the formation of charge-transfer complexes that are responsible for the fluorescence shift or quenching.11,15,42–44 However, what the formed complexes are remains controversial. For example, Dasary et al.26 reported a highly selective and ultra-sensitive, cysteine (Cys) modified gold nanoparticle based label-free surface enhanced Raman spectroscopy (SERS) probe for TNT detection at a 2 picomolar level in aqueous solution. The TNT solution varied in coloration from colorless to red when Cys was added. The authors believed the formation of a Meisenheimer7,41,45–47 complex through covalent addition of nucleophiles to a ring carbon atom of the electron-deficient aromatic substrates. The Meisenheimer complex has optical absorptions at about 520 nm and 630 nm, appearing red in solution. However, the absorption at 630 nm has not been observed by other authors for similar systems. Tu et al.15 prepared Mn2+-doped ZnS nanocrystals with a cysteamine-capping layer and used them for the fluorescence detection of ultratrace TNT by quenching their strong orange Mn2+ photoluminescence. Two absorptions at 465 nm and 515 nm were observed when cysteamine was added to the TNT solution, and the solution became red. The formation of TNT anions through an acid–base reaction was proposed. The overlap of the absorption band of the TNT anions with the emission of the Mn-doped ZnS QDs leads to the fluorescence quenching of the nano solution systems. Zou et al.42,43 synthesized Cys modified Mn2+-doped and Co2+-doped ZnS quantum dots to image ultratrace TNT in water and observed absorptions at 465 nm and 512 nm after adding TNT to the prepared QD solution. Interestingly, the authors proposed the formation of Meisenheimer complexes that quench the fluorescence emitted by the Mn2+- or Co2+-doped ZnS QDs.
There is no doubt that a complex is formed when TNT meets Cys. The complex may absorb the QDs' fluorescent emission and/or lead to the variation in color. However, there are two explanations of the mystery complex: a Meisenheimer complex through covalent bonding between the electron-deficient TNT11,48–50 and the electron-rich Cys or a TNT anion through an acid–base reaction between the acidic Cys and the basic TNT.15,43,51 Stimulated by the experimental works, we studied the interaction of molecular TNT with Cys and the structure and optical absorption of the TNT–Cys complex by means of density functional theory (DFT) calculations, aiming to make an interpretation of the experimental observations including the color change and the fluorescence quenching of TNT–Cys-QD solutions. Understanding the TNT detection rationale is certainly important to the development of its detection techniques and to the preparation of new semi-conducting QDs with proper fluorescence emissions.
2 Computational method
All computations were carried out with the Gaussian09 (ref. 52) package. First, four functionals, PBE0,53 M06-2X,54 B3LYP,55–57 and ωB97xd,58 and the 6-311++G(d,p) basis set were used to optimize the TNT structure and compute its excitation energy in acetonitrile. Cooper et al.59 measured the absorption of TNT in acetonitrile and found a strong absorption peak at 230 nm and a small absorption band over 300 nm. Our calculated results are presented in Fig. 1. Both M06-2X and ωB97xd reproduce the peak at 230 nm, while PBE0 and B3LYP underestimate the excitation energy. However, ωB97xd also predicts a strong absorption at 159 nm, which was not observed in the experiments. Therefore, M06-2X produced better results than the other three functionals and was selected in the subsequent calculations. Next, starting from the optimized TNT and Cys structures, several TNT–Cys complexes including the Meisenheimer complex were designed and optimized at the M06-2X/6-311++G(d,p) level. Frequency calculations at the same level were conducted for the lowest-energy structure to ensure that the results are true minima on the potential energy surfaces. Finally, time-dependent DFT (TDDFT) calculations were performed to simulate the optical absorptions of the Meisenheimer complex and the TNT anion and compare them with those of the experiments available. The linear response polarizable continuum model60,61 (LR-PCM) was used to mimic the solvent effect in all the calculations. The solvents were defined same as in the experiments.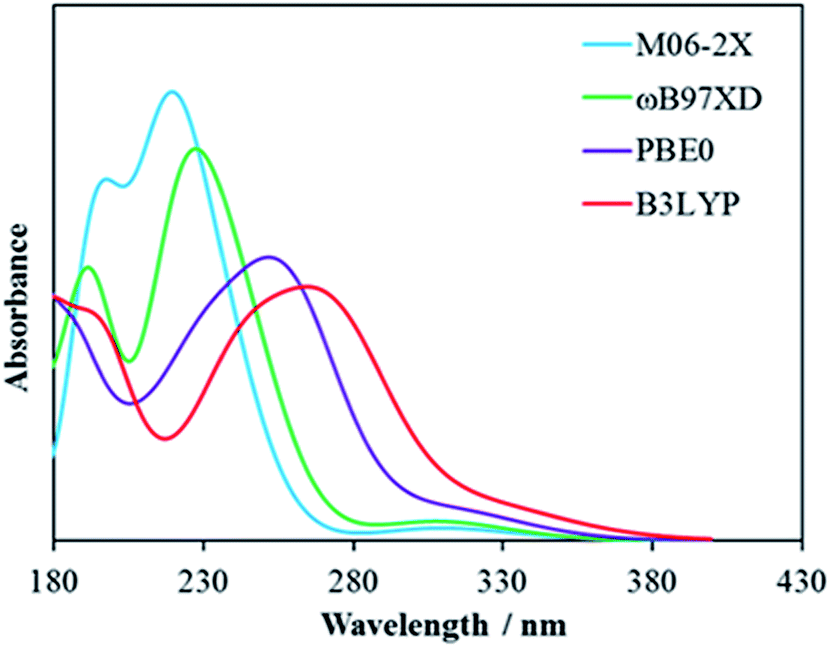 | ||
| Fig. 1 TDDFT predicted absorption spectra of TNT calculated at the M06-2X (cyan), ωB97XD (green), PBE0 (purple), and B3LYP (red) levels with the 6-311++G(d,p) basis set. | ||
3 Results and discussion
3.1 Structures of TNT–Cys complexes
Two of the designed TNT–Cys structures, the Meisenheimer complex and dehydrated TNT (S1 and S2 hereafter), were found to be considerably more stable than the others. These two structures, S1 and S2, which are also the complexes previously suggested by experimentalists,15,26,47,49,62 are formed through the reactions depicted in Scheme 1. Path I is an addition reaction in which the amine-N of Cys attacks the C1 in TNT and leads to S1. Reaction II is an acid–base reaction in which one of the methyl-H atoms is taken by the nucleophilic amine-N, and these two parts form S2 via two hydrogen bonds.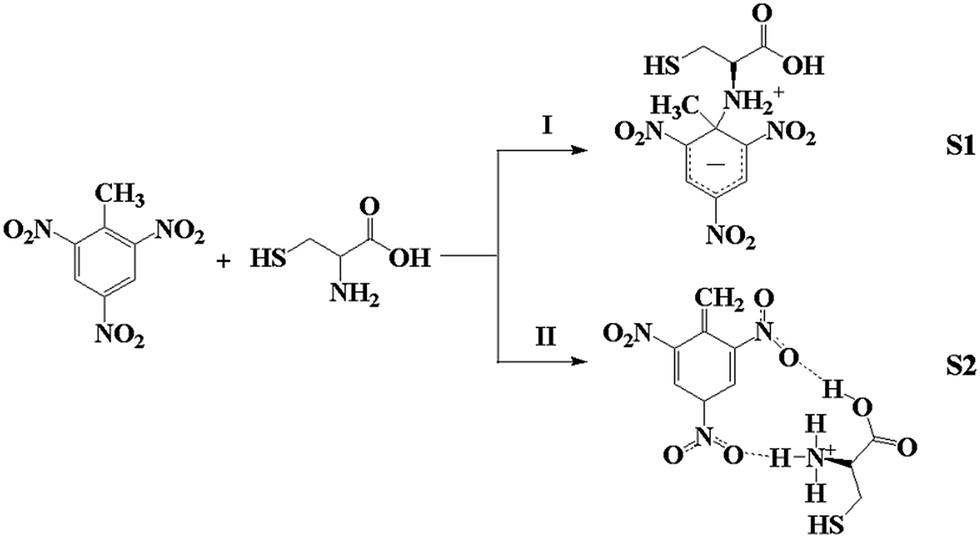 | ||
| Scheme 1 Schematic of the two reactions between TNT and Cys. | ||
The optimized structures of S1 and S2 are shown in Fig. 2, while the other less stable isomers are presented in the ESI.† Table 1 lists the selected bond lengths and bond angles of S1 and S2 obtained with M06-2X/6-311++G(d,p), as well as those of TNT and Cys for comparison. A covalent bond about 1.56 Å in length and 0.69 in bond order was predicted between Cys–N and TNT–C1, indicating a typical C–N single bond. Accordingly, the conjugated phenyl ring is partially deconstructed. C1–C2, C1–C6, and C1–C7 elongate remarkably, whereas the other C–C bonds vary by about ±0.02 Å. Comparing with the TNT structure, C1 changes from sp2 to sp3 hybridization. The conjugation path is thus blocked at C1. The bond lengths and bond angles near C1 are subjected to greater distortion than the others, as reflected by the geometrical parameters in Table 1. In addition, the amine-H is about 1.95 Å from one of the nitro-O, which is a typical HB length.
 | ||
| Fig. 2 Optimized structures of TNT, Cys, S1, S2, S3 and S4 in water. | ||
| Bond | TNT | S1 | S2 | S3 | S4 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| BL | WBO | BL | WBO | BL | WBO | BL | WBO | BL | WBO | |
| C1–C2 | 1.40 | 1.36 | 1.51 | 0.98 | 1.48 | 1.05 | 1.40 | 1.36 | 1.48 | 1.06 |
| C2–C3 | 1.38 | 1.40 | 1.36 | 1.52 | 1.37 | 1.47 | 1.39 | 1.39 | 1.37 | 1.54 |
| C3–C4 | 1.38 | 1.39 | 1.40 | 1.27 | 1.40 | 1.31 | 1.38 | 1.40 | 1.40 | 1.25 |
| C4–C5 | 1.38 | 1.39 | 1.40 | 1.27 | 1.41 | 1.23 | 1.38 | 1.37 | 1.40 | 1.25 |
| C5–C6 | 1.38 | 1.40 | 1.36 | 1.52 | 1.36 | 1.56 | 1.38 | 1.41 | 1.37 | 1.54 |
| C6–C1 | 1.40 | 1.36 | 1.52 | 1.00 | 1.48 | 1.05 | 1.40 | 1.35 | 1.48 | 1.06 |
| C1–C7 | 1.50 | 1.03 | 1.54 | 0.98 | 1.34 | 1.79 | 1.50 | 1.04 | 1.35 | 1.77 |
| C2–N | 1.48 | 0.90 | 1.43 | 1.01 | 1.42 | 1.04 | 1.48 | 0.90 | 1.44 | 0.98 |
| C4–N | 1.47 | 0.91 | 1.43 | 1.01 | 1.43 | 1.01 | 1.47 | 0.91 | 1.41 | 1.04 |
| C6–N | 1.48 | 0.90 | 1.44 | 1.00 | 1.44 | 0.98 | 1.48 | 0.89 | 1.44 | 0.98 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||
| Dihedral angle | ||||||||||
| C7–C1–C2–C3 | 174.6 | 118.3 | 150.4 | 178.3 | 161.6 | |||||
| O–N–C2–C3 | 38.3 | 9.7 | 11.1 | 35.1 | 14.1 | |||||
| O–N–C6–C5 | −38.3 | −9.4 | −26.2 | −53.5 | −14.0 | |||||
In S2 the removal of one proton from the methyl leads to a considerable change in the conjugated phenyl ring. First, the methyl-C changes from sp3 to sp2 hybridization, shortening the C1–C7 from a single bond to a double bond. C7 remains in the phenyl plane. Accordingly, the aromatic phenyl ring changes into a quinoid structure with alternating short and long C–C bonds. C2–C3 and C5–C6 are apparently shorter than C1–C2, C1–C6, C3–C4 and C4–C5.
Table 2 lists the computed energy and Gibbs free energy changes (ΔE and ΔG) of Reactions I and II. The predicted ΔE values are rather small (less than 3.4 kcal mol−1 in absolute value at the M06-2X level and 5.2 kcal mol−1 at the PBE0 level) for both reactions. The opposite signs of ΔE for Reaction I, negative (−3.3 kcal mol−1) by M06-2X and positive (4.1 kcal mol−1) by PBE0, are a reflection of their small magnitudes around zero. Both reactions have small, positive ΔG values, indicating that extra energy is required for their proceeding. Such small amounts of energy can be served either by heating or illumination in experiments. The formation of both S1 and S2 is therefore feasible under ambient conditions, although they are thermodynamically less stable than the reactants. On the other hand, S1 and S2 have similar ΔE and ΔG values (the differences are only 5.5 kcal mol−1 for ΔE and 1.9 kcal mol−1 for ΔG), indicating that they have similar thermodynamic stability. It is then possible that both S1 and S2 coexist in the system.
| Reaction | M06-2X | PBE0 | ||
|---|---|---|---|---|
| ΔE | ΔG | ΔE | ΔG | |
| I | −3.3 | 15.8 | 4.1 | 22.5 |
| II | 2.2 | 17.7 | 5.1 | 20.1 |
3.2 Optical absorption
Either S1 or S2 were considered as the key compound responsible for the fluorescence quenching of semiconducting QDs and the color change of TNT–amino-QD solutions.15,26,49,51 Starting from the optimized S1 and S2 structures, we computed their optical absorption spectra with TDDFT calculations at the M06-2X/6-311++G(d,p) level. Table 3 presents the first and second excitations and the oscillating strengths of S1 and S2.| ΔE (S0 − S1) | ΔE (S0 − S2) | |||
|---|---|---|---|---|
| λ | f | λ | f | |
| TNT | 310 | 0.017 | 237 | 0.249 |
| Cys | 203 | 0.041 | 178 | 0.016 |
| S1 | 451 | 0.219 | 340 | 0.388 |
| S2 | 508 | 0.155 | 408 | 0.366 |
| S3 | 311 | 0.010 | 277 | 0.014 |
| S4 | 523 | 0.157 | 404 | 0.348 |
First, both TNT and Cys have their absorption in the ultraviolet region, 310 nm for TNT and 203 nm for Cys, which have no effect on the color of solutions. One more possibility was considered here. TNT and Cys may form a complex via hydrogen bonds (S3 in Fig. 2). As shown in Fig. S3 in the ESI,† two HBs are formed between TNT and Cys with ΔG(298 K) = 3.5 kcal mol−1. Its predicted first excitation is at 311 nm in water. Therefore, the assembly of TNT with Cys via HBs should not be responsible for the observed fluorescence quenching or color change in the visible region either.
The computed first peak of S1 is at 451 nm, lying in the visible region. The second peak is at 340 nm, lying in the ultraviolet region. For S2, its first two peaks are at 508 nm and 408 nm. Neither S1 nor S2 have the absorption band distribution matching the observed spectra in the experiments. In Zou's50 measurement, two new bands, a strong one at 465 nm and a weak one at 516 nm, were observed when TNT was added to a Cys-modified ZnS QD aqueous solution. Similar spectra were also obtained by Tu et al.15 It is interesting to note that although the computed absorption spectra of S1 and S2 do not comply with the measurements, they possess most of the measured bands when they come together. As our calculations on the thermodynamic properties of S1 and S2 suggest the possibility of their coexistence in the solution, it is interesting to see the combination of their absorption spectra. Fig. 3 displays the optical absorption spectra of S1, S2 and their combination at a 1:1 ratio. The superposition of the spectra of S1 and S2 comes to a curve, which is very close to the observed one15,50,51,63 in band locations and shapes. The two bands at 447 nm and 506 nm are in accordance with the observed ones15,50 at about 465 nm and 515 nm.
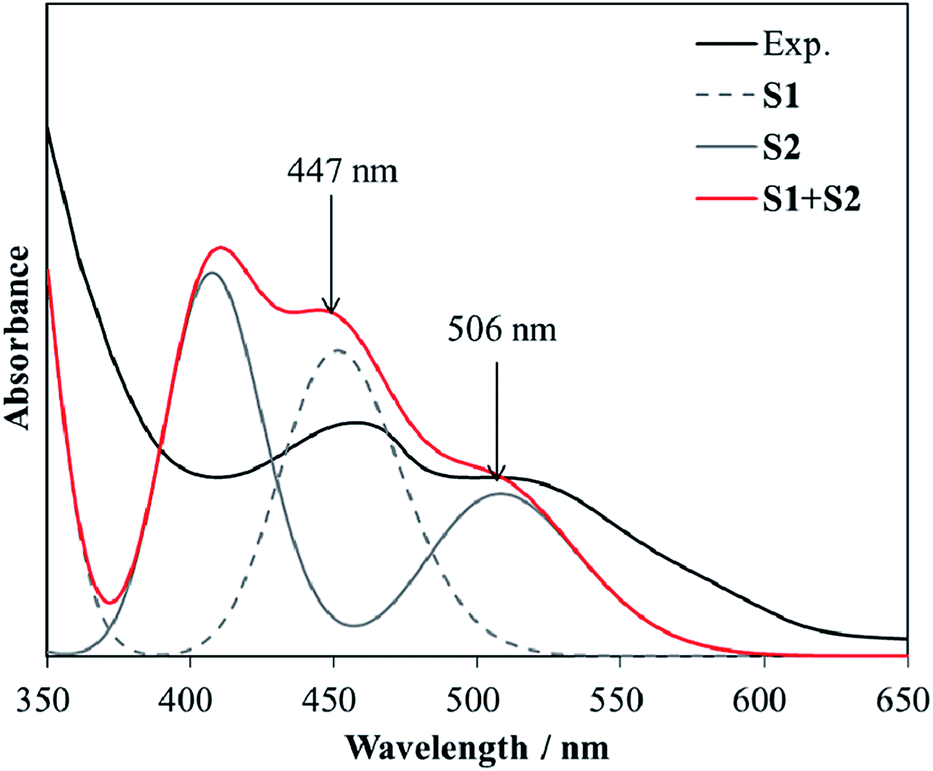 | ||
| Fig. 3 Comparison among the experimental (black line) and predicted absorption spectra of S1 (grey dashed line), S2 (grey solid line) and the combination of S1 and S2 at a 1:1 ratio (red line). | ||
Both TNT and Cys-capped QD solutions are colorless because they do not have absorption in the visible region. However, S1 and S2 are formed when these two solutions come together. S1, the Meisenheimer complex, is responsible for the absorption around 460 nm, whereas S2 is responsible for the absorption around 520 nm. This is why the mixture becomes red. The absorption and emission of Cys-capped QDs vary with the QD size, the capping ligand and the dielectric constant of the solvent. The QDs in the measurements are about 3–50 nm in diameter, with emission at 400–600 nm.15,17,42,50,51,62 It is evident that the size of QDs can be controlled to have emissions close to the absorption bands of S1 and S2. This is why the fluorescence of Cys-capped QDs is quenched in TNT solution. Our calculations revealed that the key to TNT detection is to control the QD size so as to adjust their emission to a wavelength around the absorption bands of the complexes formed with TNT and the amino acid.
In addition, we computed the optical absorption of the TNT anion (S4 in Fig. 2) in the absence of the protonated Cys. Its first two peaks are at 523 and 404 nm, which are very close to those of S2, indicating that the electron excitations in S2 are mainly contributed by the TNT anion moiety and the hydrogen bonding that binds the TNT anion and protonated Cys together has rather limited effect on the absorption spectra.
The absorption band shifts of S1 and S2 can be interpreted from their electronic structures. Our calculations revealed that the first excitations of both S1 and S2 correspond to the transitions from their highest occupied molecular orbitals (HOMO) to their lowest unoccupied molecular orbitals (LUMO). The HOMO-LUMO gaps are 5.04 eV for S1 and 4.62 eV for S2, complying with the first peak locations in their absorption spectra. Fig. 4 compares the HOMO and LUMO contours of TNT, S1 and S2. Both the HOMO and LUMO of TNT are mainly contributed by the conjugated phenyl ring, while the bonded nitro and methyl groups are also partially involved in the transition. The reaction with Cys, by either Path I or Path II, destabilizes the TNT structure and shifts its electronic excitation to longer wavelength. In S1, the conjugation in the phenyl ring was partially broken by the Cys addition. Its HOMO and LUMO are localized in the conjugated moiety. In S2, the proton removal changes the phenyl from an aromatic structure to a quinoid structure in which the conjugation path remains. Its HOMO covers all the atoms in the quinoid structure and its LUMO is also contributed from part of the atoms in the quinoid structure. The HOMO-to-LUMO transition in S2 is therefore energetically more favorable than that in S1. This is why S2 has its first absorption at a longer wavelength than S1 does. It is also revealed from Fig. 4 that the Cys part is not itself involved in the electronic excitations, but it affects the excitation by changing the electronic structure of the TNT part.
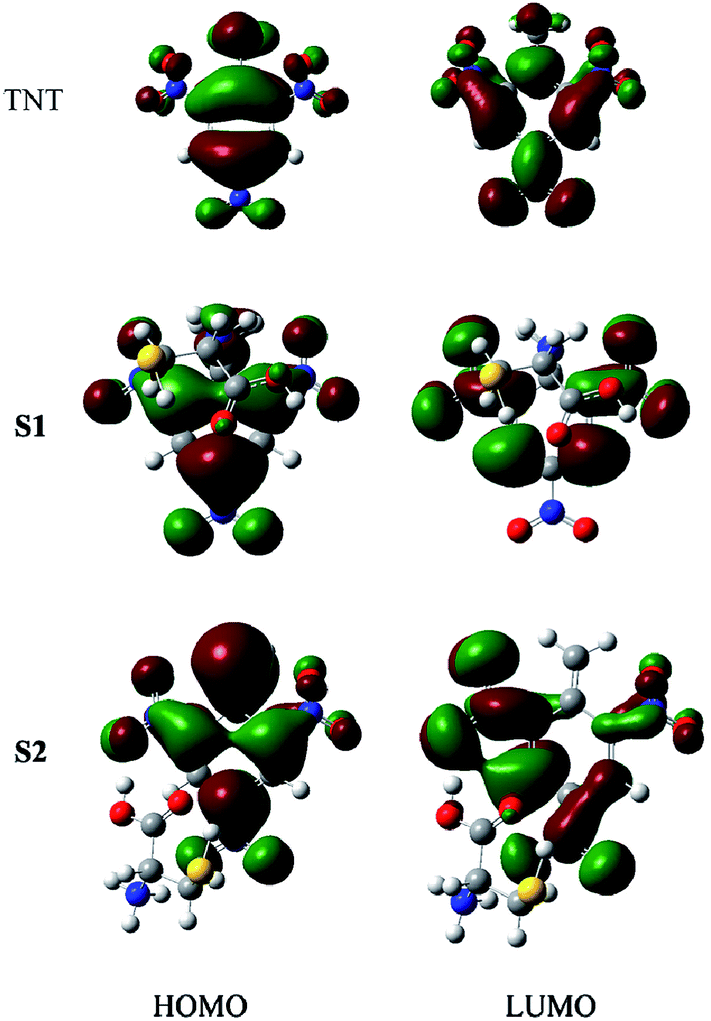 | ||
| Fig. 4 HOMO and LUMO orbitals of TNT, S1 and S2. | ||
3.3 Raman spectra
As IR and Raman spectra had been used to characterize the existence of either the Meisenheimer complex or TNT anion in a TNT–amino-QD solution,26,43,51,63 we computed the vibrational spectra with IR and Raman intensities of S1 and S2 at the M06-2X/6-311++G(d,p) level and compared them with the measurements. Fig. 5 shows the computed vibrational spectra of TNT, Cys, S1 and S2. Most of the bands of S1 and S2 can be characterized by comparing the spectrum with the TNT and Cys spectra. Affected by Cys, almost all the bands of TNT shift in the lower wavenumber direction. For example, the strongest band that is associated with NO2 symmetric stretching vibration is at 1480 cm−1 in TNT, but shifts to 1314 cm−1 in S1 and 1300 cm−1 in S2. In addition, some bands have their relative intensity and relative wave number changed significantly. Again, we take the NO2 symmetric stretching vibration as an example, which is a single band at 1300 cm−1 in S2, but split into two bands at 1460 and 1480 cm−1 in TNT, and the separation between the two bands widens to 72 cm−1 in S1.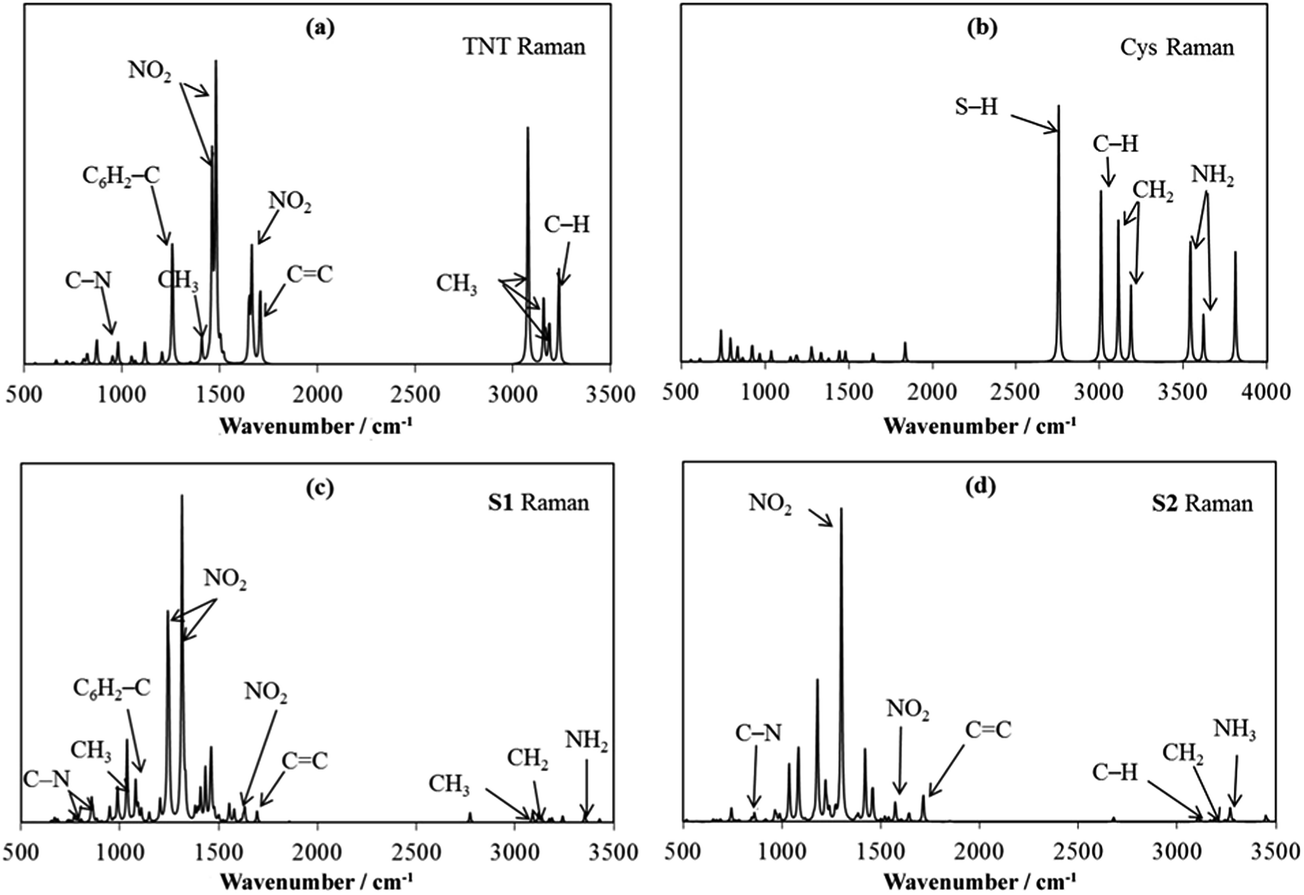 | ||
| Fig. 5 Raman spectra of TNT (a), Cys (b), S1 (c) and S2 (d). | ||
Even though significant variation in the spectra among TNT, S1 and S2 was noted, most of the bands of TNT can be traced in the S1 and S2 spectra. In addition to the characteristic peaks of the nitro groups, the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (at about 1700 cm−1), the C6H2–NO2 stretching (979 cm−1 in TNT, 989 cm−1 in S1 and 992 cm−1 in S2) and the C6H2–C stretching (1257 cm−1 in TNT and 1080 cm−1 in S1) can be clearly identified in the spectra. The strong bands in Cys are associated with C–H, N–H and S–H stretching. These bands become very weak in S1 and S2 in comparison with the strong bands of TNT.
C stretching (at about 1700 cm−1), the C6H2–NO2 stretching (979 cm−1 in TNT, 989 cm−1 in S1 and 992 cm−1 in S2) and the C6H2–C stretching (1257 cm−1 in TNT and 1080 cm−1 in S1) can be clearly identified in the spectra. The strong bands in Cys are associated with C–H, N–H and S–H stretching. These bands become very weak in S1 and S2 in comparison with the strong bands of TNT.
The formation of S1 is featured by the C1–N bond, which is at 857 cm−1 as shown in Fig. 5c. Upon its formation, the C1–CH3 bond is weakened from 1257 cm−1 to 1080 cm−1, and the N–H stretching in Cys shifts from 3542 cm−1 to 3354 cm−1. The formation of S2 is featured by the newly formed C1CH2 bond, which peaks at 1714 cm−1 and has similar vibrational frequency with the other CC stretching vibrations in the aromatic ring. The N–H stretching vibration in the newly formed NH3+ group is at 3269 cm−1.
In the Raman spectra measured by Dasary et al.,26 a strong broadband was observed at 2900 cm−1, which was assigned to the NH2 symmetric stretching, C–H stretching and CH2 asymmetric stretching in the Meisenheimer complex. The TNT peaks are at 1615 cm−1 for the CC aromatic stretching vibration, at 1360 cm−1 for the NO2 symmetric stretching vibration, 1210 cm−1 for the C1–CH3 vibration, etc. Because the measurement was conducted in the presence of gold nanoparticles, the observed bands should be to somewhat different from our calculations. The shifts are about 50–110 cm−1. However, one can still note that all the new vibrational bands featured by S1 and S2 can be associated with the observation. Both the C1–N vibration in S1 and the C1CH2 vibration in S2 can be assigned in the regions of around 850 cm−1 and around 1650 cm−1, respectively. Moreover, the broad band at 2900 cm−1 may cover the newly formed stretching vibration of NH3+ in S2. Therefore, although the observed Raman spectra did not provide adequate evidence to confirm the coexistence of S1 and S2, the observation does not exclude the possibility that both S1 and S2 are formed simultaneously when TNT meets a Cys-capped QD solution.
It should be mentioned that the effect of QDs on the structure and optical absorption of S1 and S2 was not considered in this study. The QDs are usually capped with Cys via bonding with the sulfur atom. Two saturated carbon atoms stand between the sulfur atom and the nitrogen atom that interacts with TNT. Therefore, the sulfur atom and the QD have limited influence on the electronic structures of S1 and S2. Our computations on the TNT–Cys systems are reasonable approximations for studying the mechanism of TNT detection with amino-capped QDs.
4. Conclusion
In order to reveal the mechanism of TNT detection with amino acid-capped QDs, DFT calculations were performed to study the structures and optical properties of the products of TNT reacting with Cys. A number of candidate TNT–Cys complex structures were designed and optimized at the DFT level. A Meisenheimer complex (S1) and TNT anion (S2) were identified as the two most stable structures among the candidates. In previous experimental studies, either S1 or S2 were identified as the product. However, our TDDFT computations for either S1 or S2 failed to reproduce the measured spectra. Instead, the superposition of their absorption bands matches well with the measurements. The coexistence of these two complexes was thus suggested and further confirmed in terms of their similar thermodynamic stability and comparisons between predicted and measured Raman spectra. The color change and fluorescence quenching were then interpreted with the optical absorption of both S1 and S2. The absorption bands of S1 and S2 are at 451 and 508 nm, respectively, which can be matched by the emission of QDs. Our calculations revealed that the key to TNT detection is to control the size of the QDs so as to adjust their emission to a wavelength around the absorption bands of the complexes formed with TNT and amino acid.Acknowledgements
The authors thank financial support from the National Natural Science Foundation of China (no. 21373140) and Supported by National High Technology Research and Development Program of China (no. 2015AA034202). Part of calculations was carried out at the State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University.Notes and references
- W. D. Won, L. H. Disalvo and J. Ng, Appl. Environ. Microbiol., 1976, 31, 576–580 CAS.
- R. Hernandez, M. Zappi and C. H. Kuo, Environ. Sci. Technol., 2004, 38, 5157–5163 CrossRef CAS.
- P. van Dillewijn, J. L. Couselo, E. Corredoira, A. Delgado, R. M. Wittich, A. Ballester and J. L. Ramos, Environ. Sci. Technol., 2008, 42, 7405–7410 CrossRef CAS.
- O. J. Hao, K. K. Phull, A. P. Davis, J. M. Chen and S. W. Maloney, Water Environ. Res., 1993, 65, 213–220 CrossRef CAS.
- S. Singh, J. Hazard. Mater., 2007, 144, 15–28 CrossRef CAS PubMed.
- M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC.
- R. Ban, F. F. Zheng and J. R. Zhang, Anal. Methods, 2015, 7, 1732–1737 RSC.
- W. J. Qi, M. Xu, L. Pang, Z. Y. Liu, W. Zhang, S. Majeed and G. B. Xu, Chem.–Eur. J., 2014, 20, 4829–4835 CrossRef CAS PubMed.
- B. X. Liu, C. Y. Tong, L. J. Feng, C. Y. Wang, Y. He and C. L. Lu, Chem.–Eur. J., 2014, 20, 2132–2137 CrossRef CAS PubMed.
- H. X. Zhang, L. J. Feng, B. X. Liu, C. Y. Tong and C. L. Lu, Dyes Pigm., 2014, 101, 122–129 CrossRef CAS PubMed.
- L. J. Feng, C. Y. Tong, Y. He, B. X. Liu, C. Y. Wang, J. Sha and C. L. Lu, J. Lumin., 2014, 146, 502–507 CrossRef CAS PubMed.
- S. F. Xu and H. Z. Lu, Chem. Commun., 2015, 51, 3200–3203 RSC.
- A. Rose, Z. G. Zhu, C. F. Madigan, T. M. Swager and V. Bulovic, Nature, 2005, 434, 876–879 CrossRef CAS PubMed.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- R. Y. Tu, B. H. Liu, Z. Y. Wang, D. M. Gao, F. Wang, Q. L. Fang and Z. P. Zhang, Anal. Chem., 2008, 80, 3458–3465 CrossRef CAS PubMed.
- W. Wei, X. B. Huang, K. Y. Chen, Y. M. Tao and X. Z. Tang, RSC Adv., 2012, 2, 3765–3771 RSC.
- L. J. Feng, H. Li, Y. Qu and C. L. Lu, Chem. Commun., 2012, 48, 4633–4635 RSC.
- R. Freeman, T. Finder, L. Bahshi, R. Gill and I. Willner, Adv. Mater., 2012, 24, 6416–6421 CrossRef CAS PubMed.
- N. Niamnont, N. Kimpitak, K. Wongravee, P. Rashatasakhon, K. K. Baldridge, J. S. Siegel and M. Sukwattanasinitt, Chem. Commun., 2013, 49, 780–782 RSC.
- Y. R. Wang, Y. X. Gao, L. Chen, Y. Y. Fu, D. D. Zhu, Q. G. He, H. M. Cao, J. G. Cheng, R. S. Zhang, S. Q. Zheng and S. M. Yan, RSC Adv., 2015, 5, 4853–4860 RSC.
- L. Liu, J. Y. Hao, Y. T. Shi, J. S. Qiu and C. Hao, RSC Adv., 2015, 5, 3045–3053 RSC.
- C. L. Zhang, K. J. Wang, D. J. Han and Q. Pang, Spectrochim. Acta, Part A, 2014, 122, 387–391 CrossRef CAS PubMed.
- X. He, H. Wang, Z. B. Li, D. Chen and Q. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 14706–14712 RSC.
- A. K. M. Jamil, E. L. Izake, A. Sivanesan and P. M. Fredericks, Talanta, 2015, 134, 732–738 CrossRef CAS PubMed.
- H. X. Gu, L. Xue, Y. H. Zhang, Y. F. Zhang and L. Y. Cao, Adv. Mater. Res., 2014, 924, 366–370 CrossRef CAS.
- S. S. R. Dasary, A. K. Singh, D. Senapati, H. T. Yu and P. C. Ray, J. Am. Chem. Soc., 2009, 131, 13806–13812 CrossRef CAS PubMed.
- E. M. A. Ali, H. G. M. Edwards and I. J. Scowen, Talanta, 2009, 78, 1201–1203 CrossRef CAS PubMed.
- D. D. Tuschel, A. V. Mikhonin, B. E. Lemoff and S. A. Asher, Appl. Spectrosc., 2010, 64, 425–432 CrossRef CAS PubMed.
- H. B. Zhou, Z. P. Zhang, C. L. Jiang, G. J. Guan, K. Zhang, Q. S. Mei, R. Y. Liu and S. H. Wang, Anal. Chem., 2011, 83, 6913–6917 CrossRef CAS PubMed.
- Z. Z. Guo, A. Florea, C. Cristea, F. Bessueille, F. Vocanson, F. Goutaland, A. D. Zhang, R. Sandulescu, F. Lagarde and N. Jaffrezic-Renault, Sens. Actuators, B, 2015, 207, 960–966 CrossRef CAS PubMed.
- M. C. Casey and D. E. Cliffel, Anal. Chem., 2015, 87, 334–337 CrossRef CAS PubMed.
- L. L. Zhang, Y. J. Han, J. B. Zhu, Y. L. Zhai and S. J. Dong, Anal. Chem., 2015, 87, 2033–2036 CrossRef CAS PubMed.
- Y. X. Gan, R. H. Yazawa, J. L. Smith, J. C. Oxley, G. Zhang, J. Canino, J. Ying, G. Kagan and L. H. Zhang, Mater. Chem. Phys., 2014, 143, 1431–1439 CrossRef CAS PubMed.
- M. Riskin, R. Tel-Vered, T. Bourenko, E. Granot and I. Willner, J. Am. Chem. Soc., 2008, 130, 9726–9733 CrossRef CAS PubMed.
- X. C. Fu, X. Chen, J. Wang, J. H. Liu and X. J. Huang, Electrochim. Acta, 2010, 56, 102–107 CrossRef CAS PubMed.
- M. Y. Ho, N. D'Souza and P. Migliorato, Anal. Chem., 2012, 84, 4245–4247 CrossRef CAS PubMed.
- N. Hebestreit, J. Hofmann, U. Rammelt and W. Plieth, Electrochim. Acta, 2003, 48, 1779–1788 CrossRef CAS.
- C. Carrillo-Carrion, B. M. Simonet and M. Valcarcel, Anal. Chim. Acta, 2013, 792, 93–100 CrossRef CAS PubMed.
- T. Pazhanivel, D. Nataraj, V. P. Devarajan, V. Mageshwari, K. Senthil and D. Soundararajan, Anal. Methods, 2013, 5, 910–916 RSC.
- L. J. Feng, C. Y. Wang, Z. L. Ma and C. L. Lu, Dyes Pigm., 2013, 97, 84–91 CrossRef CAS PubMed.
- E. Buncel, J. M. Dust and F. Terrier, Chem. Rev., 1995, 95, 2261–2280 CrossRef CAS.
- W. S. Zou, D. Sheng, X. Ge, J. Q. Qiao and H. Z. Lian, Anal. Chem., 2011, 83, 30–37 CrossRef CAS PubMed.
- W. S. Zou, J. Q. Qiao, X. Hu, X. Ge and H. Z. Lian, Anal. Chim. Acta, 2011, 708, 134–140 CrossRef CAS PubMed.
- W. S. Zou, F. H. Zou, Q. Shao, J. Zhang, Y. Q. Wang, F. Z. Xie and Y. Ding, J. Photochem. Photobiol., A, 2014, 278, 82–88 CrossRef CAS PubMed.
- A. Haidour and J. L. Ramos, Environ. Sci. Technol., 1996, 30, 2365–2370 CrossRef CAS.
- P. G. Rieger, V. Sinnwell, A. Preuss, W. Francke and H. J. Knackmuss, J. Bacteriol., 1999, 181, 1189–1195 CAS.
- F. Fant, A. De Sloovere, K. Matthijsen, C. Marle, S. El Fantroussi and W. Verstraete, Environ. Pollut., 2001, 111, 503–507 CrossRef CAS.
- Y. F. Chen, Z. Chen, Y. J. He, H. L. Lin, P. T. Sheng, C. B. Liu, S. L. Luo and Q. Y. Cai, Nanotechnology, 2010, 21, 125502 CrossRef PubMed.
- K. Zhang, H. B. Zhou, Q. S. Mei, S. H. Wang, G. J. Guan, R. Y. Liu, J. Zhang and Z. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8424–8427 CrossRef CAS PubMed.
- W. S. Zou, J. Yang, T. T. Yang, X. Hu and H. Z. Lian, J. Mater. Chem., 2012, 22, 4720–4727 RSC.
- Y. Q. Wang and W. S. Zou, Talanta, 2011, 85, 469–475 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.01, Gaussian Inc., Wallingford Ct, 2009 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. K. Cooper, C. D. Grant and J. Z. Zhang, J. Phys. Chem. A, 2013, 117, 6043–6051 CrossRef CAS PubMed.
- R. Cammi, M. Cossi and J. Tomasi, J. Chem. Phys., 1996, 104, 4611–4620 CrossRef CAS PubMed.
- R. Cammi, M. Cossi, B. Mennucci and J. Tomasi, J. Chem. Phys., 1996, 105, 10556–10564 CrossRef CAS PubMed.
- S. F. Xu, H. Z. Lu, J. H. Li, X. L. Song, A. X. Wang, L. X. Chen and S. B. Han, ACS Appl. Mater. Interface, 2013, 5, 8146–8154 CrossRef CAS PubMed.
- W. S. Zou, Y. Q. Wang, F. Wang, Q. Shao, J. Zhang and J. Liu, Anal. Bioanal. Chem., 2013, 405, 4905–4912 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07088k |
| This journal is © The Royal Society of Chemistry 2015 |