DOI:
10.1039/C5RA07074K
(Paper)
RSC Adv., 2015,
5, 47390-47397
Size-dependent physicochemical properties of mesoporous nanosilica produced from natural quartz sand using three different methods
Received
19th April 2015
, Accepted 18th May 2015
First published on 20th May 2015
Abstract
Mesoporous high-surface-area silica (SiO2) nanoparticles were produced from natural quartz sand (orthoquartzite) using three processing methods namely sol–gel, sonication, and spray pyrolysis. The inexpensive precursor was extracted from the quartz sand by alkali extraction followed by acid precipitation, which was used for all the three methods. The effects of production methods were investigated by various characterization techniques. The physicochemical properties of the obtained nanoparticles were compared to explore the effect of size and porosity on their electronic, optical, mechanical, and electrical qualities. The produced SiO2 nanoparticles were found to have an amorphous high surface area in the range of 178–322 m2 g−1 and a uniform size distribution with the high purity and spherical morphology. These particles formed a mesoporous material with an average pore diameter of 10–26 nm. It was found that the surface area (178 < 284 < 322 m2 g−1) and band gap (5.41 < 5.43 < 5.45 eV) of the particles increased with a decrease in particle size (39 > 30 > 10 nm) when the process method was changed from sol–gel to sonication and from sonication to spray pyrolysis. This study provides useful insights and guidance for the preparation of mesoporous SiO2 nanoparticles from quartz sand and throws light on how physicochemical properties are influenced by process methods and particle size.
1. Introduction
The production of amorphous high-surface-area silica (SiO2) nanoparticles has drawn great interest due to their wide range of potential applications in electronic devices, insulators, catalysis, enzyme encapsulation, drug delivery, and agriculture.1–9 The emerging new technological innovations demand different forms of silica such as precipitated silica, powder silica, fumed silica, silica gel, and silica sol. Generally, precursors such as tetraethyl orthosilicate (TEOS), silicon alkoxide, and sodium silicate are used for the production of silica. The commercial preparation of silicon alkoxides from raw silica sand involves a multistep reaction route of carbothermal reaction at approximately 1300 °C.10,11 Both the above processes are highly expensive and associated with high temperature, strong acidity, high pressure as well as eco-hazards. The sharp increase in porous silica applications demands the development of an economically viable method to fabricate nano-silica from abundant silicon-containing natural materials such as quartz sand and biomass rice husk.12–15
Alkali extraction and fluorination methods are reportedly used to recover the silica from quartz sand.11,12 Some of the general methods used to produce silica particles are precipitation and hydrolysis,16 sol–gel,17 sonication,18 stober process,19 ball-milling,20 thermal decomposition,21 wet chemical,22 microemulsion,23 spray pyrolysis,24 and vapor phase hydrolysis.25 Using the well-established procedures, nano-silica (nano-SiO2) particles with uniform size, shape, purity, and high surface area are prepared for different industrial applications.
The knowledge on the process parameters plays a key role in determining the size, shape, structural morphologies, and surface area of SiO2 nanoparticles. Sol–gel process is considered to be an excellent method to synthesize various nanometal oxides with desired size, structure, and morphology.17 In sonication method, ultrasound irradiation is used for the synthesis of mesoporous materials with reduced time and aggregation of nanoparticles into porous structures without destroying their micellar structure.18 Similarly, in the atomized spray pyrolysis technique the choice of precursors, concentration, droplet size, and temperature is used to form ultrafine and uniform ceramic powders.24 Sol–gel, sonication, and spray pyrolysis are the most common and significant methods for the production of SiO2 nanoparticles with controlled size and morphology. These are very simple, cost-effective, and energy-efficient methods that are well suited for mass production. The establishment of relationship between the physicochemical properties and requires information on uniform particle size, surface area, and purity. Among these parameters, particle size and porosity play an important role in determining the properties.17 The review of literature indicates that the mass production of high purity nano-SiO2 from natural minerals sand for the industrial applications is found to be scanty.
The main objective of this study was to synthesize amorphous high-surface-area SiO2 nanoparticles from natural mineral (quartz sand) through an inexpensive chemical process. To explore the effect of the change in the size, shape, and morphology of SiO2 nanoparticles for different industrial applications, an attempt has been made in the present study to produce SiO2 nanoparticles with size ranging from 10 to 50 nm using three methods, namely sol–gel, sonication, and spray pyrolysis. The obtained nanoparticles and their properties were compared to explore the effect of size and porosity on their electronic, optical, mechanical, and electrical qualities. The correlation established between these will be more beneficial for effective use of the natural mineral (quartz sand) to produce mass quantities of nano-SiO2 to meet the requirements of industrial applications.
2. Experimental sections
2.1 Materials
The quartz sand was collected from Indian Peninsula (from southern part of eastern Tuticorin). The chemical composition of quartz sand is given in Table 1. The chemical reagents such as hydrochloric acid (HCl; 37 wt%), sodium hydroxide pellets (NaOH; 98 wt%), and sulfuric acid (H2SO4; 98 wt%) were purchased from Merck (India) and used without any further purification.
Table 1 Chemical composition of natural quartz sand
| Quartz sand |
Composition |
SiO2 |
K2O |
P2O5 |
Fe2O3 |
CaO |
| Weight (%) |
66.48 |
19.79 |
10.26 |
1.61 |
1.46 |
2.2 Sample pretreatment and precursor preparation
Yellow, irregularly shaped quartz sand with a rough surface and particle size of ∼100–200 μm were crushed by planetary ball mill (PM 100; Retsch, Germany) at 300 rpm to reduce the particle size range to few micrometers. The initial reduction in the particle size helps to enhance the chemical reactions. The sand was washed thoroughly using 35 wt% of HCl solutions twice. The obtained sand was washed repeatedly with double-distilled (DD) water to remove free transition metal ions. Then, the sand was filtered to leach out other metal salts in water. The change in color of the powder from yellow to mild white confirmed the effectiveness of the HCl treatment in removing the transition metal ions and trace metals. The filtrated pure silicate residue was used for an alkali leaching via dry fusion method. An equal amount of silicate residue and sodium hydroxide pellet was mixed together and fused in a muffle furnace at 400–450 °C for 3 h under atmospheric air. Then, the mixture containing water-soluble sodium silicate was leached with DD water. The solution was filtrated using a filter paper (Whatman, UK) to remove the unreacted silicate residue, and the process was repeated twice to obtain a clear and colorless solution. The fusion reaction of silicate (quartz sand) with NaOH pellet is shown in the following reaction:| | |
SiO2(quartz sand) + NaOH → Na2SiO3 + residue
| (1) |
2.3 Synthesis of SiO2 nanoparticle using sol–gel method
To the extracted sodium silicate precursor (250 ml) 10 N of H2SO4 was added drop wise under constant stirring till a pH 9 was achieved. During this reaction, hydrolysis and condensation take place in the sodium silicate precursor and finally nano-silica gel was obtained. The silica nanoparticles were obtained as follows:| | |
Na2SiO3 + H2SO4 → SiO2(gel) + Na2SO4 + H2O
| (2) |
The gel thus obtained was washed with DD water (both normal and boiled) to remove alkali/sodium sulfate impurities. It was then digested at 80 °C for 32 h with controlled gel nucleation followed by drying at 120 °C for 3 h in a hot-air oven to obtain solid silica. The solid was ground into powder using a plenary ball mill at 400 rpm for 2 h to avoid agglomeration of particles. Hereafter, the nano-SiO2 sample prepared from sol–gel method is termed as S1 sample. Further, S1 sample was calcined at 400 °C for 3 h in a muffle furnace.
2.4 Synthesis of SiO2 nanoparticle using sonication method
To the precursor solution, 10 N of H2SO4 was drop wise added until it reaches the pH 9 and the gel was formed under sonication (VC 505, Sonics, USA) with a constant ultrasound irradiation (35 kHz). The gel was diluted initially with DD water and then ultrasound irradiation was performed within the diluted solution under ambient air for 3 h at regular time interval. After sonication, the obtained solution of hydrous SiO2 was washed with DD water and then dried and calcined at ambient conditions. The obtained sonicated nano-SiO2 sample is hereafter termed as S2 sample.
2.5 Synthesis of amorphous high-surface-area SiO2 nanoparticle using spray pyrolysis method
The SiO2 gel was precipitated at pH 9 by titrating with 10 N of H2SO4 in sodium silicate precursor. The gel was washed with DD water to obtain pure silica solution and then to form a transparent solution with DD water. It was used as the starting phase in spray pyrolysis. In the spray pyrolysis method, reaction often takes place in solution droplets, followed by solvent evaporation. The transparent solution was drop wise added to the chamber through a nozzle along with pressurized compressed air for atomization, as shown in Fig. 1. Atomizer formation was controlled by the pressure of compressed air and the flow rate of precursors was controlled by the feed pump. The sprayed and atomized nano-sized entities of SiO2 were decomposed at 400 °C in the reaction chamber to obtain SiO2 nanoparticles. After the completion of one full cycle, the produced SiO2 nanoparticles were collected from the cyclones and hereafter termed as S3 sample. Further, S3 sample was calcined at 400 °C for 3 h in a muffle furnace.
 |
| | Fig. 1 Flow diagram of the process methods used to convert quartz sand into silica nanoparticles. | |
2.6 Characterization
The obtained SiO2 powder sample was characterized using comprehensive characteristic techniques. Amorphous/crystalline phases of all powder samples were detected through powder X-ray diffraction (XRD) patterns (X'Pert PRO; PANalytical, the Netherlands) using CuKα as a radiation source (λ = 1.54060 Å). The diffractometer was operated at 30 kV and scanned from 10° to 80° of 2θ, at a scanning rate of 10° per min. Fourier transform infrared (FTIR) measurement was carried out by preparing pellet at room temperature in the frequency range of 4000–400 cm−1 using a spectrometer (Spectrum 100; PerkinElmer, USA). The powder samples were mixed with KBr in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. The pellet was prepared by subjecting it to a load of 5 tons cm−3. The silica powder was directly suspended in DD water with brief sonication and the particle size distribution was measured using dynamic light-scattering technique (Nanophox; Sympatec, Germany). Qualitative and quantitative elemental analyses of powder were carried out using an energy-dispersive spectrometer (JED-2300; JEOL, Japan) and a X-ray fluorescence (XRF) spectrometer (XGT-2700; Horiba, Japan).
:100. The pellet was prepared by subjecting it to a load of 5 tons cm−3. The silica powder was directly suspended in DD water with brief sonication and the particle size distribution was measured using dynamic light-scattering technique (Nanophox; Sympatec, Germany). Qualitative and quantitative elemental analyses of powder were carried out using an energy-dispersive spectrometer (JED-2300; JEOL, Japan) and a X-ray fluorescence (XRF) spectrometer (XGT-2700; Horiba, Japan).
Scanning electron microscope (SEM; JSM-6390LV; JEOL, Japan) was used to analyze the surface morphology of the samples. Transmission electron microscopy (TEM; CM200; Philips, Netherlands) was used to study the shape and size of the particles. The adsorption–desorption isotherms were carried out for all the prepared samples in liquid N2 at −196 °C using a Brunauer–Emmett–Teller (BET) surface area analyzer (Autosorb AS-1-MP; Quantachrome, FL). It was used to determine the specific surface area, that is, SSABET (m2 g−1), pore volume, and pore radius with the help of BET equation:26
| |
 | (3) |
where
V is the volume of nitrogen (cm
3 g
−1) absorbed at equilibrium pressure,
P the saturation vapor pressure (
P0) at absolute temperature
T,
Vm the monolayer capacity (cm
3 g
−1), and
C the BET constant. Solid-state ultraviolet-visible (UV-Vis) absorption spectra were recorded using a UV-Vis diode array spectrometer (8453, Agilent, Singapore). The bandgap energy (
Eg) of the SiO
2 samples was obtained using the following energy equation:
| |
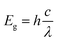 | (4) |
where
Eg is the bandgap energy (eV),
h the Planck's constant,
c the velocity of light and
λ the wavelength. The photoluminescence (PL) spectral measurements were carried out using a spectrometer (FP-6500; JASCO, UK).
The samples were characterized by X-ray photoelectron spectroscopy (XPS) to reveal the chemical composition and the relative concentration of silica and oxygen on the nano-SiO2 particles using XPS spectrometer (PHOIBOS HSA3500, SPECS, Germany). The spectra were recorded using a monochromatic source of AlKα (1486.74). The analysis was done at an ambient temperature (300 K) at a vacuum level of less than 4 × 10−10 Torr. Structure of the nano silica was analyzed on a NMR spectrometer (Mercury Plus 300 MHz, Varian, USA) operating at 59.6 MHz for solid state 29Si. The samples were spun at a spinning rate of 5 kHz. The mechanical test for all the prepared samples was conducted using the nano-indentation Triboindenter (quasistatic nanoindentation; T1 700 Ubi; Hysitron, USA). The indentation was carried out for the prepared pellet during each loading and unloading at a time interval of 5 s with a maximum force of 600 μN and a scanning area of 10 × 10 μm. The hardness and elastic modulus of nanoparticles were obtained using the standard relationship between the applied indentations load (P) and measured penetration depth (h) using the following relation:27
| |
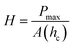 | (5) |
| |
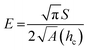 | (6) |
where
P is the applied maximum force,
A(
hc) the area of the function,
A the area,
hc the contact depth, and
S the contact stiffness from slope of loading and unloading cure.
3. Results and discussion
The XRD patterns of samples S1, S2, and S3 are shown in Fig. 2. The absorbed patterns for all three samples are similar and confirm the amorphous phase of silica with the absence of sharp intense peaks and high-intensity broad diffraction peak at 22.5°. The result is also supported by similar pattern for silica obtained from rice husk combustion.28 The FTIR spectral assignments of samples S1, S2, and S3 are shown in Fig. 3. The absorbance of peak at 470 cm−1 corresponds to the Si–O–Si stretching vibration.29 Further, the peaks observed at 800 and 1630 cm−1 correspond to the hydroxyl group Si–OH.29 The broad absorbance peaks observed between 1016 and 1248 cm−1 correspond to the Si–O stretching vibration.29 It can be concluded from the FTIR studies that all the prepared samples confirm the existence of Si and O.
 |
| | Fig. 2 XRD patterns of nano SiO2 samples S1, S2 and S3. | |
 |
| | Fig. 3 FTIR spectra of nano SiO2 samples S1, S2 and S3. | |
The particle size distribution of S1, S2, and S3 samples is shown in Fig. 4. It is in the range of 26 (d10)–54 (d90), 13 (d10)–59 (d90), and 4 (d10)–32 (d90) nm size, respectively, for S1, S2, and S3 samples. Moreover, the mean size distribution (d50) of particles is 39 ± 3, 30 ± 3, and 10 ± 3 nm, respectively, for samples S1, S2, and S3. S1 and S3 samples yielded narrow size distributed particles whereas S2 sample resulted in wide size distributed particles.
 |
| | Fig. 4 Particle size distribution of nano SiO2 samples dispersed in ethanol. | |
This may be due to the method used for the production of SiO2 nanoparticles wherein the breakage of newly formed silica particles leads to disintegration and aggregation of the particles.24 Whereas, both mean particle size and particle distribution are reduced in spray pyrolysis method when compared to other two methods (i.e., sol–gel and sonication). The elemental analysis and chemical composition of SiO2 nanosamples are shown in Table 2. All the three samples are yield 98 wt% of SiO2. Further, it is evident from the XRF analysis (Table 2) that all the produced samples comprise 99 wt% of SiO2.
Table 2 EDX and XRF analysis of samples
| Samples |
EDX Analysis |
XRF Analysis |
| Composition |
Mass (%) |
Composition |
Weight (%) |
| S1 |
SiO2 |
98.72 |
SiO2 |
99 |
| S2 |
SiO2 |
98.69 |
SiO2 |
99 |
| S3 |
SiO2 |
98.82 |
SiO2 |
99 |
The isotherm curves of S1, S2, and S3 are shown in Fig. 5a and c. SSABET of as-produced S1, S2, and S3 are, respectively, 178 ± 10, 284 ± 10, and 322 ± 10 m2 g−1. The obtained SSABET results are in good agreement with those of particle size analysis and are compared in Table 3. Moreover, it can be seen that the SSABET of the particle increases with a decrease in particle size. The reduced particle size in turn increases the surface excess atoms due to the increase in surface-to-volume ratio. The hysteresis loop appears between the adsorption and desorption branches. The entire isotherm belongs to type V category based on the IUPAC classification, and the absorbed hysteresis loop shows close resemblance with typical pattern of mesoporous materials based on Brunauer classification.30,31
 |
| | Fig. 5 BET results of porous nano SiO2 samples (a) adsorption–desorption isotherm (b) pore size distribution, (c) 7-point BET plot. | |
Table 3 Particle size, BET and nano indentation analysis of samples
| Samples |
Particle size nm |
Particle size distribution nm |
BET surface area m2 g−1 |
Band gap energy eV |
Hardness GPa |
Elastic modulus GPa |
| S1 |
39 ± 3 |
26–54 |
178 ± 10 |
5.41 |
2.02 |
20.89 |
| S2 |
30 ± 3 |
13–59 |
284 ± 10 |
5.43 |
1.13 |
12.64 |
| S3 |
10 ± 3 |
4–28 |
322 ± 10 |
5.46 |
0.72 |
8.20 |
The capillary condensation and evaporation does not take place at the same relative pressure, hence it may lead to the appearance of hysteresis loop in S1 and S3 samples but in the S2 samples having small loop. The existence of mixture of hysteresis loop between type H2 and H3 indicates relatively high pore size uniformity and facile pore connectivity.31 The obtained SSABET, average pore diameter, and particle size ranges are given in Table 3 for comparison. The isotherm represents the mesoporosity (2–50 nm in pore size) in the silica according to IUPAC definition for mesoporosity.32 According to the Barrett–Joyner–Halenda (BJH) method, the pore size distribution is calculated in the present study and the results are shown in Fig. 5b. The capillary condensation leading to a sharp increase in absorbed volume commences at lower relative pressure in S3 (P/P0 = 0.57) than S1 (P/P0 = 0.8). Thus, it leads to a wider pore size distribution. Therefore, S1 and S3 have different adsorption and desorption behaviors. On the other hand, the particle size and surface area of S3 is higher than S1 leading to lower pore diameter in S3 than S1.32 The S1 and S3 have different adsorption and desorption behavior. But the particle size and surface area is higher for S3 when compared to S1, this may be the reason to have lower pore diameter for S3 when compare to S1 pore diameter. The observed results show that the SSABET and mesoporosity increase the pore size, which in turn decreases the pore volume due to the effect of synthesis methods.
Fig. 6a–c shows the SEM image of as-synthesized nano-SiO2 samples. The obtained SiO2 has a weak agglomerated surface morphology and its size ranges from several tens to hundred nanometers. It is evident from the figure that S1 and S2 samples yield weak agglomerated particles with irregular morphology whereas S3 sample yields weakly agglomerated particles with spherical morphology. The TEM analysis further confirms the spherical morphology as observed in the SEM images. The spot rings in the selected area diffraction (SAED) pattern are indexed as amorphous SiO2. The observed SAED pattern matches well with the XRD pattern (Fig. 2). The TEM micrographs reveal that the distribution of prepared SiO2 nanoparticle for samples S1, S2, and S3 is in the size range of 30–45, 20–35, and 10–25 nm, respectively. The absorbed TEM images (Fig. 6d–f) confirm that the nanoparticles prepared using three methods resemble with the obtained particle size distribution (Fig. 4).
 |
| | Fig. 6 SEM and TEM micrographs of nano SiO2 samples (a) SEM image of S1 (b) SEM image of S2 (c) SEM image of S3 (d) TEM image of S1 (e) TEM image of S2 (f) TEM image of S3. | |
The full XPS survey spectrum of sol–gel SiO2 (S1) sample is shown in Fig. 7. The electron core level XPS spectra of SiO2 namely Si2p, Si2s, O1s, O2s, C1s and Sc indicate the presence of silicon and oxygen components in the sample. The absorbed Si2p signal at 103.5 eV (Fig. 7a and c) reveals the existence of pure SiO2.33–36 The absorbed Si2s signal peak at 154.2 eV (Fig. 7a and d) corresponds to the presence of Si species in the sample.34 The O1s spectra signal peak absorbed at 532.3 eV (Fig. 7a and e) clearly indicates that higher contribution of oxygen with porous silica.33–36 The O2s signal peak at 24.0 eV (Fig. 7a and b) reveals the presence of oxygen species in the sample.34 A small hump near 286 eV and 496 eV indicates respectively the presence of residual carbon and scandium atoms in the sample, which is may due to the process contamination and the source material quartz sand.33–36 The above observation shows that the sample S1, S2 and S3 contains highly pure SiO2 components. The absorbed XPS results are in close resemblance with the EDAX and XRF results as given in Table 2. Solid state 29Si NMR spectra of samples are shown in Fig. 8. The 29Si NMR shifts are known to exist in the range between +50 and −200 ppm, and a maximum peak is absorbed at −108.5 ppm. It reveals a symmetric shape of the peak attributed to Q4[Si(OSi)4] tetrahedral phase found in amorphous silica.36–38 The high intensity of Q4 signal indicates a large amount of Si atoms are fully condensed with the other Si atoms and oxygen atoms.36–38 The 29Si NMR spectra results further confirms the existence of Si–O–Si structure as obtained from FTIR structure (Fig. 3).
 |
| | Fig. 7 XPS scanning spectra of nano-SiO2 sample (a) XPS full survey spectrum (b) magnified O2s spectrum (c) magnified Si2p spectrum (d) magnified Si2s spectrum (e) magnified O1s spectrum. | |
 |
| | Fig. 8 29Si NMR spectra of nano-SiO2 sample. | |
The optical absorption spectra measured by the UV-Vis spectrometer for all three methods used to obtain nano-SiO2 samples are shown in Fig. 8. The onset wavelength (λmax) is obtained by extrapolating the absorption edge along x axis (Fig. 8). The absorption values (λmax) for samples S1, S2, and S3 are 229, 228, and 227 nm, respectively. The bandgap energy of the amorphous nano-SiO2 samples is calculated using the eqn (4). The bandgap energy of samples S1, S2, and S3 is found to be 5.41 eV (229 nm), 5.43 eV (228 nm), and 5.46 eV (227 nm), respectively. The bandgap energy (Eg) values are in close agreement with those of size of SiO2 nanoparticles, as reported earlier.39,40 The particle size decreases from 39 (S1) to 30 (S2) nm and from 30 (S2) to 10 (S3) nm with an increase in bandgap energy value from 5.41 (S1) to 5.43 (S2) eV and from 5.43 (S2) to 5.46 (S3) eV. The SiO2 nanoparticles show absorption curve shifts to the lower wavelength (blueshift) when the particle size is decreased from ∼40 to 10 nm. The peak position observed at ∼229 nm (5.4 eV) in Fig. 9 is attributed to the stabilization of E′ centers, which increase linearly with increasing particle size.40
 |
| | Fig. 9 UV-Vis spectra of nano SiO2 samples S1, S2 and S3. | |
Fig. 10 shows the typical PL spectra with the excitation wavelength of ∼400 nm. It can be seen that all SiO2 samples show obvious excitonic PL signals with a similar curve shape. It is used to frame the discussion that the production methodology, particle size, and specific surface area can alter the excitonic wavelength and show PL emission at 394–401 nm. Slight blueshift is observed for the band intensity of nano-SiO2 samples relative to the size and is influenced by Si–O species through intra- and interparticle interactions. Thus, it can be concluded that the increase in intensity of the band and excitonic wavelength confirms the blueshift with decrease in particle size of the SiO2 nanoparticles, as reported earlier.39,40 Therefore, the observed string emission in the region of 390–410 nm is assigned to the emission originating from the blending of a hole with an electron.
 |
| | Fig. 10 PL spectra of nano SiO2 samples S1, S2 and S3. | |
It can be observed that the PL intensity significantly depends on particle size and surface area of SiO2 nanoparticles. The average experimental data between indentation force and penetration depth curves (load and displacement) for the nano-SiO2 samples prepared using three methods are shown in Fig. 11. The elastic modulus of the nano-SiO2 is found to be 20, 12, and 8 GPa for samples S1, S2, and S3, respectively. The modulus of nano-SiO2 samples is 5–10 times lower than the reported value approximately 72 GPa for bulk fused silica.40–43 The hardness (H) value of the as-prepared nano-SiO2 samples is in the range between 2 and 0.7 GPa. It is evident from the above results that the elastic modulus and hardness values of the samples decrease as the particle size decreases, which is observed due to the increase in the surface area and porosity of the nanoparticles, as reported elsewhere.44,45
 |
| | Fig. 11 Typical load–displacement curves of nano SiO2 samples S1, S2 and S3. | |
4. Conclusions
In this study, a natural silica sources (quartz sand) was used to synthesize porous SiO2 nanoparticles by reducing the number of chemical modifications and energy consumptions. The high-surface-area SiO2 nanoparticles with a diameter range of 10–40 nm were successfully prepared from quartz sand (orthoquartzite) via alkaline extraction followed by three methods (namely, sol–gel, sonication, and spray pyrolysis). The electronic, optical, mechanical, and electrical properties of the prepared SiO2 nanoparticles were studied through different characterization studies. It can be concluded that the spray pyrolysis is an effective method to produce the required SiO2 nanoparticles. The resultant SiO2 nanoparticles were intensively studied to determine their size-dependent physicochemical and mechanical properties. Overall, significant enhancements in the physicochemical properties were achieved with the decrease in particle size, as the particle size decreased in proportion to the hardness. Also, decreased hardness was found to be more sensitive to the surface area values and particle size. Therefore, the prepared SiO2 nanoparticles can be used for industrial applications such as gas adsorptions, molecular separation, catalysis and automobiles, and in drug delivery and biomedical applications.
Acknowledgements
One of the authors Mr S. Arunmetha is thankful for the Senior Research Fellowship (SRF) grant by Council of Scientific and Industrial Research (CSIR), New Delhi (sanction no. 08/570(0003)/2014-EMR-I dt. 07.08.2014) to carry out this research work. The authors are also thankful to Dr G. Amarendra and Dr Shamima Hussain, UGC-DAE Consortium for Scientific Research, Kalpakkam Node, for the XPS spectra measurements.
Notes and references
- C. Gerardin, J. Reboul, M. Bonne and B. Lebeau, Chem. Soc. Rev., 2013, 42, 4217 RSC.
- D. Tahir, H. L. Kwon, H. C. Shin, S. K. Oh, H. J. Kang, S. Heo, J. G. Chung, J. C. Lee and S. Tougaard, J. Phys. D: Appl. Phys., 2010, 43, 255301 CrossRef.
- M. G. Veena, N. M. Renukappa, J. M. Raj, C. Ranganathaiah and K. N. Shivakumar, J. Appl. Polym. Sci., 2011, 121, 2752 CrossRef CAS PubMed.
- T. Gao, B. P. Jelle, L. I. C. Sandberg and A. Gustavsen, ACS Appl. Mater. Interfaces, 2013, 5, 761 CAS.
- X. Li, Y. Yang and Q. Yang, J. Mater. Chem. A, 2013, 1, 1525 CAS.
- N.-T. Chen, S.-H. Cheng, J. S. Souris, C.-T. Chen, C.-Y. Mou and L.-W. Lo, J. Mater. Chem. B, 2013, 1, 3128 RSC.
- H. A. A. Wab, K. A. Razak and N. D. Zakaria, J. Nanopart. Res., 2014, 16, 2256 CrossRef.
- X. Huang, N. P. Young and H. E. Townley, Nanomater. Nanotechnol., 2014, 4, 21 Search PubMed.
- R. Suriyaprabha, G. Karunakaran, R. Yuvakkumar, P. Prabu, V. Rajendran and N. Kannan, J. Nanopart. Res., 2012, 14, 1294 CrossRef.
- X. Chen, J. Jiang, F. Yan, S. Tian and K. Li, RSC Adv., 2014, 4, 8703 RSC.
- R. K. Iler, The Chemistry of Silica, John Wiley & Sons, New York, 1979 Search PubMed.
- L. P. Demyanova, V. Rimkevich and A. S. Buynovskiy, J. Fluorine Chem., 2011, 132, 1067 CrossRef CAS PubMed.
- H. Chen, W. Wang, J. C. Martin, A. J. Oliphant, P. A. Doerr, J. F. Xu, K. M. DeBorn, C. Chen and L. Sun, ACS Sustainable Chem. Eng., 2013, 1, 254 CrossRef CAS.
- D. P. Wong, R. Suriyaprabha, R. Yuvakumar, V. Rajendran, Y.-T. Chen, B.-J. Hwang, L.-C. Chen and K.-H. Chen, J. Mater. Chem. A, 2014, 2, 13437 CAS.
- R. Yuvakkumar, V. Elango, V. Rajendran and N. Kannan, J. Exp. Nanosci., 2014, 9, 272–281 CrossRef CAS PubMed.
- K. D. Kim and H. T. Kim, J. Sol-Gel Sci. Technol., 2002, 25, 183 CrossRef CAS.
- I. A. Rahman and V. Padavettan, J. Nanomater., 2012, 15, 132424 Search PubMed.
- N. Enomoto, T. Koyano and Z.-e. Nakagawa, Ultrason. Sonochem., 1996, 3, S105 CrossRef CAS.
- T. Gholami, M. S. Niasari, M. Bazarganipour and E. Noori, Superlattices Microstruct., 2013, 61, 33 CrossRef CAS PubMed.
- M. S. Niasari, J. Javidi and M. Dadkhah, Comb. Chem. High Throughput Screening, 2013, 16, 458 CrossRef.
- P. N. R. Kishore and P. Jeevanandam, J. Alloys Compd., 2012, 522, 51 CrossRef CAS PubMed.
- R. Stanley and A. S. Nesaraj, Int. J. Appl. Sci. Eng., 2014, 12, 9 Search PubMed.
- K. S. Finnie, J. R. Bartlett, C. J. A. Barbe and L. Kong, Langmuir, 2007, 23, 3017 CrossRef CAS PubMed.
- A. G. Howard and N. H. Khdary, Mater. Lett., 2007, 61, 1951 CrossRef CAS PubMed.
- X. Chen, J. Jiang, F. Yan, S. Tian and K. Li, RSC Adv., 2014, 4, 8703 RSC.
- A. F. Hassan, A. M. Abdelghny, H. Elhadidy and A. M. Youssef, J. Sol-Gel Sci. Technol., 2014, 69, 65 CrossRef.
- O. Sahin, O. Uzun, M. S. Lizer, H. Gocmez and U. Kölemen, J. Eur. Ceram. Soc., 2008, 28, 1235 CrossRef CAS PubMed.
- P. K. Jal, M. Sudarshan, A. Saha, S. Patel and B. K. Mishra, Colloids Surf., A, 2004, 240, 173 CrossRef CAS PubMed.
- M. Noushadn, I. A. Rahman, N. S. C. Zulkifli, A. Husein and D. Mohamad, Ceram. Int., 2014, 40, 4163 CrossRef PubMed.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723 CrossRef CAS.
- K. S. W. Sing, Reporting physisorption data for gas/solid systems, Pure Appl. Chem., 1982, 54(11), 2201 CrossRef.
- M. Jafarzadeh, I. A. Rahman and C. S. Sipaut, J. Sol-Gel Sci. Technol., 2009, 50, 328 CrossRef CAS PubMed.
- D. S. Jensen, S. S. Kanyal, N. Madaan, M. A. Vail, A. E. Dadson, M. H. Engelhard and M. R. Linford, Surf. Sci. Spectra, 2013, 20, 36 CrossRef.
- J. Chastain, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, 1992 Search PubMed.
- N. Pal, E.-B. Cho and D. Kim, RSC Adv., 2014, 4, 9213 RSC.
- J. Kanungo, L. Selegard, C. Vahlberg, K. Uvdal, H. Saha and S. Basu, Bull. Mater. Sci., 2010, 33, 647 CrossRef CAS PubMed.
- E.-B. Cho, S. Yim, D. Kim and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 12595 CAS.
- V. V. Brei, J. Chem. Soc., Faraday Trans., 1994, 90, 2961 RSC.
- M. Jafarzadeh, I. A. Rahman and C. S. Sipaut, Ceram. Int., 2010, 36, 333 CrossRef CAS PubMed.
- I. A. Rahman, P. Vejayakumaran, C. S. Sipaut, J. Ismail and C. K. Chee, Mater. Chem. Phys., 2009, 114, 328 CrossRef CAS PubMed.
- J. Malzbender, J. M. J. den Toonder, A. R. Balkenende and G. D. With, Mater. Sci. Eng., R, 2002, 36, 47 CrossRef.
- C. P. Wong and R. S. Bollampally, J. Appl. Polym. Sci., 1999, 74, 3396 CrossRef CAS.
- B. Bhushan, Handbook of Nanotechnology, Springer, Berlin, 2004 Search PubMed.
- L. Zhang, M. D. Acunzi, M. Kappl, G. K. Auernhammer and D. Vollmer, Langmuir, 2009, 25, 2711 CrossRef CAS PubMed.
- J. Yin, M. Retsch, J.-H. Lee, E. L. Thomas and M. C. Boyce, Langmuir, 2011, 27, 10492 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 














