
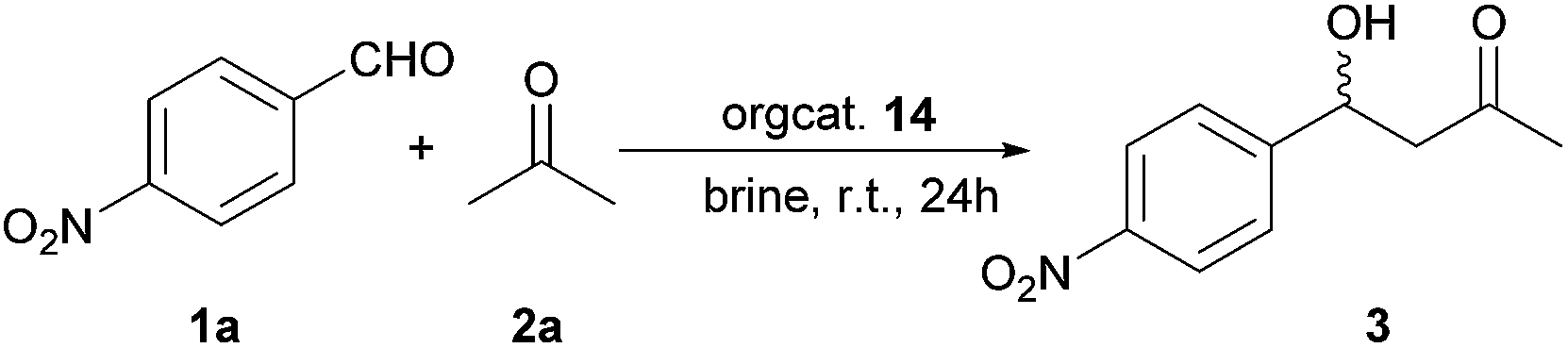
Highly modular dipeptide-like organocatalysts for direct asymmetric aldol reactions in brine†
Xiao-Mu Hu,
Dong-Xu Zhang,
Sheng-Yong Zhang* and
Ping-An Wang*
Department of Medicinal Chemistry, School of Pharmacy, The Fourth Military Medical University, Changle West Road 169, Xi'an, 710032, P. R. China. E-mail: ping_an1718@outlook.com; syzhang@fmmu.edu.cn; Fax: +86-29-84776945; Tel: +86-29-84776807 ext. 605
First published on 24th April 2015
Abstract
A novel series of dipeptide-like organocatalysts derived from proline, amino acids and primary amines have been prepared for direct asymmetric aldol reactions between various aromatic aldehydes and acetone to afford aldol products in good yields (up to 82%) and moderate enantioselectivities (up to 67% ee) with only 1 mol% of catalyst-loading in brine. Under the same conditions, the direct asymmetric aldol reactions of aromatic aldehydes and cyclohexanone give aldol products with high yields (up to 91%) and moderate to good enantioselectivities (up to 88% ee) and excellent diastereoselectivities (up to 99% dr). These organocatalysts are easily synthesized from commercially available materials in multi-gram scale with high modularity in their structural and stereogenic properties.
Introduction
The direct aldol reaction is one of the most fundamental reactions for carbon–carbon bond formation.1,2 Many efforts are devoted to develop a highly efficient asymmetric version of this important reaction both in metal3 or metal-free catalytic conditions.4 Since the prominent achievements of List and Barbas III on proline catalysis,5 a large number of organocatalysts have been prepared and applied in direct aldol reactions between various aldehydes and ketones to achieve excellent yields and enantioselectivities.6–19Peptides are common bio-molecules and constituents in living organisms, and play vital roles in variously biological activities. In modern organic synthesis, small peptides can be used as catalysts and ligands for enantioselective transformations20 such as acylation,21 epoxidation,22 and cyclopropanation.23 Undoubtedly, the research of organic synthesis in or on aqueous medium is attractive to chemists by its environmentally benign property and easily handled process.24 Since Janda's25 pioneering work on direct asymmetric aldol reactions in water, Barbas III,26 Hayashi,27 and Singh28 et al. have independently developed enantioselective aldol reactions in aqueous media to achieve excellent results. Small peptides are also utilized as organocatalysts for direct asymmetric aldol reactions in water or brine to afford aldols in good yields and enantioselectivities,29 and some elegant examples30 are listed in Fig. 1. However, the high catalyst-loading (usually 10–30 mol%) and additives are necessary to ensure good yields and enantioselectivities in most cases, and the low-loading organocatalysts without any additive for the direct asymmetric aldol reactions under aqueous conditions are rare.
 | ||
| Fig. 1 Typical small peptides catalysed direct aldol reactions. | ||
In this paper, we have presented the preparation and application of a novel class of highly modular dipeptide-like organocatalysts derived from proline, amino acids and primary amines for direct aldol reactions between various aromatic aldehydes and ketones in brine to provide aldol products in excellent yields and moderate to good enantioselectivities with only 1 mol% of catalyst-loading (Fig. 2). Different amino acid tethered proline-skeletons were compared and their catalytic behaviour was also evaluated in this study.
 | ||
| Fig. 2 Dipeptide-like organocatalysts in this study. | ||
Results and discussion
Synthesis of dipeptide-like organocatalysts
The synthesis of these dipeptide-like organocatalysts is very straightforward and shown in Scheme 1. The coupling of the chiral (or achiral) primary amines 9 with different Boc-protected amino acids 10 under the mixed-anhydride conditions provides the intermediates 11 in high yields which directly followed the deprotection of Boc-group to afford the amines 12. The connection of 12 with Boc-proline can produce 13 which undergo the deprotection to yield the final dipeptide-like organocatalysts 14 (Fig. 3). Only one column chromatographic purification of the intermediates 13 needed in the third step. The organocatalysts 14b–e derived from L-proline and (S)-methylbenzylamine but containing different amino acid motifs in the middle part of the structures, are prepared for the detection of the steric effect on their chiral induction abilities in the direct aldol reactions. The family of 14f–h are synthesized from chiral methylbenzylamine, valine and proline but with different configurations to investigate the effect of the tunably stereogenic centers on aldol reactions. The organocatalyst 14i with the ending group of benzylamine is used in the direct aldol reaction to show the necessity of the chiral or achiral primary amine group in these dipeptide-like analogues. The catalyst 14a [from L-proline and (S)-methylbenzylamine] are used as catalysts for the comparison with the performance of the above dipeptide-like organocatalysts 14b–i in the direct aldol reactions. | ||
| Scheme 1 The synthetic route to 14c. | ||
 | ||
| Fig. 3 The structures of organocatalysts 14a–i. | ||
Direct aldol reactions by using dipeptide-like organocatalysts
With these above dipeptide-like organocatalysts 14b–i in hand, the direct aldol reaction of 4-nitrobenzyaldehyde and acetone was chosen as the model reaction for the test of the activities of catalysts. Initially, the reaction was performed under neat conditions at room temperature (r.t.) with catalysts L-proline (30 mol%), and 14a–e (5 mol% respectively), and the results are shown in the Table 1. From the Table 1, it was found that L-proline was the effective catalyst to give moderate yield (76%) and relatively good enantioselectivity (67% ee) of aldol product (Table 1, entry 1) but with a large amount of catalyst-loading. The catalyst 14a with slightly positive result of aldol reaction has good yield (87%) but with very low enantioselectivity (Table 1, entry 2, 20% ee). The catalyst 14b from L-proline, L-alanine and (S)-methylbenzylamine (L-Pro-L-Ala-S-amine) gives 82% yield and 37% ee of aldol product (Table 1, entry 3). With the increase of the steric hindrance of the middle part of the catalysts, the yield and the enantioselectivity of aldol adduct is also significantly increased (Table 1, entries 3–5). To our surprise, the most hindered t-Bu in the middle part of catalyst 14d affords lower yield and enantioselectivity than the catalyst 14c (L-Pro-L-Val-S-amine, LLS, Table 1, entry 4 vs. 5). The catalyst 14e with L-Phe in the middle part results the lower enantioselectivity than 14c (Table 1, entry 6 vs. 4). When the reaction is performed at −40 °C in neat acetone, the yield of aldol product is decreased but with the increase of the enantioselectivity (Table 1, entry 4 with 14c, 75% ee vs. 57% ee) and the sacrifice of reaction time (up to 3 days).
|
|||||
|---|---|---|---|---|---|
| Entrya,b | Orgcat. | r.t.b | −40 °Cc | ||
| Yield (%) | ee (%)d | Yield (%) | ee (%)d | ||
| a 0.5 mmol of aldehyde 1a in 1.0 mL of acetone for every entry.b The reaction time is 24 h.c The reaction time is 3 days.d The enantiomeric excess (ee) of aldol product was determined by chiral HPLC with Chiralpak AS-H column.e 30 mol% of L-proline was used in entry 1. | |||||
| 1e | L-Pro | 76 | 67(R) | — | — |
| 2 | 14a | 87 | 20(R) | 67 | 39(R) |
| 3 | 14b | 82 | 37(R) | 62 | 59(R) |
| 4 | 14c | 92 | 57(R) | 68 | 75(R) |
| 5 | 14d | 84 | 45(R) | 65 | 61(R) |
| 6 | 14e | 84 | 41(R) | 67 | 57(R) |
| 7 | 14f | 92 | 55(R) | 63 | 80(R) |
| 8 | 14g | 94 | 57(S) | 57 | 53(S) |
| 9 | 14h | 93 | 57(S) | 61 | 49(S) |
| 10 | 14i | 90 | 49(R) | 59 | 47(R) |

In order to investigate the effect of the tunable stereogenic centers of catalysts on aldol reactions, 14f (L-Pro-L-Val-R-amine, LLR), 14g (D-Pro-D-Val-S-amine, DDS), 14h (D-Pro-D-Val-R-amine, DDR) and 14i (L-Pro-L-Val-Bn, LLBn) were prepared respectively. The direct aldol reactions of 4-nitrobenzyaldehyde and acetone under neat condition at r.t. or at −40 °C were performed by using these above catalysts. It was found that the configuration of the chiral carbon atom in the terminal amine of catalysts is changed from (S)- to (R)-, the enantioselectivity of aldol product is slightly increased from 75% (r.t., with catalyst 14c) to 80% ee (−40 °C, with catalyst 14f, Table 1, entry 4 vs. 7). Switching the configuration of proline motif of catalysts from L- to D-, the configurations of aldol product are also changed to be their opposite configurations (Table 1, entries 4, 7 vs. 8, 9). When the terminal amine is achiral (catalyst 14i), the enantioselectivity of aldol product is decreased (Table 1, entry 10). These results indicate that the different stereogenic centers in these dipeptide-like catalysts can produce the obviously different enantioselective inductions and 14f is the best choice both at r.t. and −40 °C on the direct aldol reactions.
To the environmental benign concern, the direct aldol reaction was undertaken in brine at r.t. with the catalysts 14c, 14f–i. There is a decrease of at least 10% ee for every entry when changing from neat conditions to brine (Table 2, entries 1–5). The catalyst-loading is also investigated. Interestingly, the enantioselectivity of aldol product is obviously decreased to be 23% ee with 10 mol% of catalyst-loading of 14f (Table 2, entry 6 vs. entry 2), and this may due to the reaction switching from kinetic-control to thermodynamic-control.31 To our delight, when the amount of 14f is reduced to be 1 mol%, the aldol product is afforded in slightly lower yield (80%) but with relatively stable enantioselectivity (45% ee) comparison to 5 mol% of catalyst-loading (Table 2, entry 7 vs. entry 2). However, when the reaction was performed in water with 1 mol% of catalyst 14f, the enantioselectivity is decreased dramatically to 7.5% ee (Table 2, entry 8 vs. entry 7). This shows that these dipeptide-like organocatalysts are highly efficient for the direct aldol reactions in brine but with very poor performance in water.
|
|||
|---|---|---|---|
| Entrya | Orgcat. (mol%) | Yield (%) | ee (%)b |
| a 0.5 mmol of aldehyde 1a and 0.5 mL of acetone in 1.0 mL of brine for every entry.b The enantiomeric excess (ee) of aldol product was determined by chiral HPLC with Chiralpak AS-H column, the configuration of aldol product was assigned by comparison to the literature data.c The reaction was performed in water. | |||
| 1 | 14c(5) | 79 | 44(R) |
| 2 | 14f(5) | 80 | 45(R) |
| 3 | 14g(5) | 83 | 45(S) |
| 4 | 14h(5) | 82 | 45(S) |
| 5 | 14i(5) | 89 | 35(R) |
| 6 | 14f(10) | 85 | 23(R) |
| 7 | 14f(1) | 80 | 45(R) |
| 8c | 14f(1) | 77 | 7.5(R) |
By using only 1 mol% of dipeptide-like organocatalyst 14f, the direct aldol reactions of acetone and various aromatic aldehydes (Table 3) with electron-withdrawing or electro-donating group are suitable to brine medium at r.t. to produce aldol adducts in good yields (up to 82%) and moderate enantioselectivities (up to 67% ee).
|
||||
|---|---|---|---|---|
| Entrya | Substrate | Product | Yield (%) | ee (%)b |
| a 0.5 mmol of aldehyde 1 and 0.5 mL of acetone in 0.5 mL of brine for every entry.b The enantiomeric excess (ee) of aldol products 3a–j was determined by chiral HPLC with Chiralpak AD-H, OD-H and AS-H columns, the configuration of aldol product was assigned by comparison to the literature data. | ||||
| 1 | 1a R = 4-NO2 | 3a | 80 | 45(R) |
| 2 | 1b R = 2-NO2 | 3b | 82 | 60(R) |
| 3 | 1c R = 3-NO2 | 3c | 80 | 56(R) |
| 4 | 1d R = 4-Br | 3d | 75 | 51(R) |
| 5 | 1e R = 3-Br | 3e | 78 | 52(R) |
| 6 | 1f R = H | 3f | 60 | 54(R) |
| 7 | 1g R = 2,4-dichloro | 3g | 67 | 41(S) |
| 8 | 1h R = 4-Me | 3h | 61 | 54(R) |
| 9 | 1i R = 3-OMe | 3i | 65 | 67(R) |
| 10 | 1j R = 3,4,5-(OMe)3 | 3j | 78 | 47(R) |
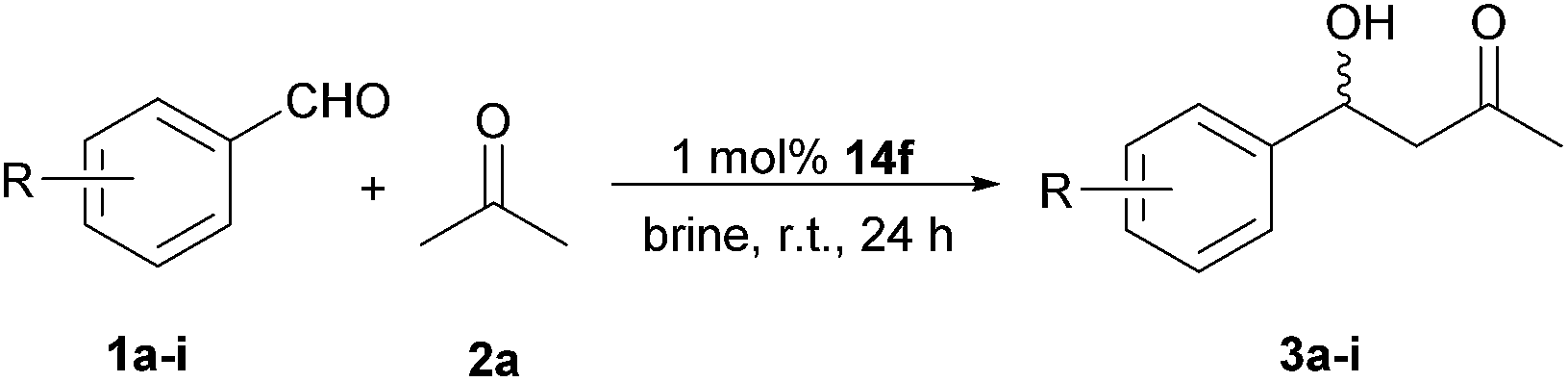
The direct aldol reaction of 4-nitrobenzyaldehyde and cyclohexanone (Table 4, entry 1) in water with 1 mol% of 14f afforded 15a in excellent diastereoselectivity (d.r. > 98%) and moderate enantioselectivity of anti-product (54% ee). However, switching the reaction solvent of water to brine, the enantioselectivity of 15a is significantly increased to 88% ee (Table 4, entry 2). The direct aldol reactions between various aromatic aldehydes 1a–k and cyclohexanone in brine with 1 mol% of catalyst 14f were smoothly performed at r.t. to furnish aldols 15a–k with moderate to good enantioselectivities (except benzaldehyde 1f, entry 7, 28% ee) and excellent diastereoselective ratios (Table 4, entries 2–12).
|
|||||
|---|---|---|---|---|---|
| Entrya | Substrate | Product | Yield (%) | Anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :syn :syn |
ee (%)d |
| a 0.5 mmol of aldehyde 1 and 0.5 mL of acetone in 0.5 mL of brine for entries 2 to 12.b The reaction was performed in 0.5 mL of acetone with 0.5 mL of water for entry 1.c For entry 12, the substrate 1k is 4-chlorobenzyaldehyde.d The enantiomeric excess (ee) of aldol products 15a–k was determined by chiral HPLC with Chiralpak AD-H column, the configuration of aldol product was assigned by comparison to the literature data. | |||||
| 1b | 1a | 15a | 81 | 98:2 |
54 |
| 2 | 1a | 15a | 82 | 96:4 |
88 |
| 3 | 1b | 15b | 89 | 99:1 |
68 |
| 4 | 1c | 15c | 92 | 99:1 |
65 |
| 5 | 1d | 15d | 85 | 98:2 |
77 |
| 6 | 1e | 15e | 86 | 99:1 |
76 |
| 7 | 1f | 15f | 72 | 98:2 |
28 |
| 8 | 1g | 15g | 87 | 99:1 |
78 |
| 9 | 1h | 15h | 76 | 95:5 |
84 |
| 10 | 1i | 15i | 81 | 97:3 |
67 |
| 11 | 1j | 15j | 80 | 89:11 |
66 |
| 12c | 1k | 15k | 91 | 91:9 |
81 |

From the above reaction results, we have noticed that not only acetone but also cyclohexanone react with various aromatic aldehydes in brine to afford aldol adducts with superior enantioselectivities to those in water (Table 2, entry 7 vs. entry 8; Table 4, entry 2 vs. entry 1). This observation may due to the “salting-out effect” increasing “hydrophobic effect” which was found in the previous reports by Barbas III,26 Hayashi27 and Singh28 et al. The OH groups of the surface water molecules in brine may form the proper amount of additional hydrogen bonds with two amide oxygen atoms to make the amidic NH more acidic resulting in a tight transition state in hydrophobic micro-surroundings to ensure the good enantioselectivity. When the aldol reaction was performed in water, a large amount of hydrogen bonds might form not only with two amide oxygen atoms but also with amidic NH groups to deteriorate the stable transition state leading to poor enantioseletivity. The proposed transition states are shown in Fig. 4.
 | ||
| Fig. 4 The proposed transition states in brine and in water. | ||
In summary, we have prepared a novel series of dipeptide-like organocatalysts for the direct asymmetric aldol reactions of various aromatic aldehydes and acetone in brine with only 1 mol% of catalyst-loading to provide aldol products in good yields (up to 82%) and moderate enantioselectivities (up to 67% ee). Additionally, under the same reaction conditions, the direct asymmetric aldol reactions of various aromatic aldehydes with cyclohexanone afford the corresponding aldol adducts in high yields (up to 92%) and moderate to good enantioselectivities (up to 88% ee) with excellent diastereoselectivitives (d.r. up to 99%). These dipeptide-like organocatalysts are easily synthesized in multi-gram scale with high tunability in their structural and stereogenic properties. The more complex dipeptide-like organocatalysts with multi-hydrogen bonding sites are investigated in due course.
Experimental
General
Melting points are uncorrected and expressed in °C by MRS-2 melting point apparatus from Shanghai Apparatus Co., Ltd. 1H NMR and 13C NMR spectra were measured in CDCl3, solution on a Bruker AV-400 spectrometer using TMS as an internal reference. Coupling constant (J) values are given in Hz. Multiplicities are designated by the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; br, broad; m, multiplet. High-resolution mass spectra were performed on a Bruker microTOF-Q II Mass Spectrometer with ES ionization (ESI). All commercially available reagents were used as received. Thin-layer chromatography on silica (with GF254) was used to monitor all reactions. Products were purified by flash column chromatography on silica gel purchased from Qingdao Haiyang Chemical Co., Ltd. Optical rotations were measured on a Perkin Elmer 343 polarimeter. Chiral High Performance Liquid Chromatography (HPLC) analyses were performed using an Agilent 1200 series apparatus and Chiralpak AD-H, OD-H and AS-H columns purchased from Daicel Chemical Industries. The configuration of the products has been assigned by comparison to the literature data or assigned by analogy. All reactions involving air- or moisture-sensitive species were performed in oven-dried Schlenk tubes under an inert atmosphere.Typical synthetic procedure for preparation of dipeptide-like organocatalyst 14c
A solution of Boc-L-Val-OH (10c, 2.17 g, 10 mmol), Et3N (1.52 g, 15 mmol) and isobutyl chloroformate (1.57 mL, 10 mmol) in dichloromethane (DCM, 25 mL) at 0 °C was stirred for 30 min, after which the (S)-(−)-1-phenylethylamine 9 (11 mmol) was added and stirred at this temperature for 2 h after which it was warmed to room temperature along with stirring overnight. The reaction was monitored by TLC, and then quenched with sat. aq. NH4Cl (25 mL), and successively washed with H2O, and brine. Aqueous phase extracted with dichloromethane (3 × 25 mL). The organic layers were dried over Na2SO4, filtered, and concentrated to give the crude 11c as light yellow glue which was directly deprotected by using trifluoroacetic acid (TFA, 4 mL, 52 mmol, 6 equiv.) in 30 mL of DCM to afford the crude product 12c without further purification. The coupling of Boc-L-Pro-OH (1.72 g, 8 mmol) with crude 12c was performed following the same procedure of the connection of 10c with 9 to yield the crude 13c as yellow wax (Alternatively, the crude 13c can be purified by a flash column chromatography with n-hexane/ethyl acetate = 1/1, V/V). This crude 13c was directly undergone the deprotection of Boc-group via TFA (4 mL, 52 mmol) in anhydrous DCM (30 mL) produced the crude product 14c which was purified by a flash column chromatography (n-hexane/ethyl acetate/Et3N = 1/5/0.05, V/V) to provide the final dipeptide-like organocatalyst 14c (1.87 g, total yield 59% based on Boc-L-Val-OH).(S)-N-((S)-1-phenylethyl)pyrrolidine-2-carboxamide32 (14a)
Compound 14a was prepared from Boc-L-Pro-OH and (S)-(−)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used.Yield 72%; white solid; m.p. 145–147 °C; [α]25D = −91.8° (c = 0.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 7.2 Hz, 1H), 7.36–7.34 (m, 5H), 5.11 (dd, J = 7.2, 1.6 Hz, 1H), 3.75 (dd, J = 5.6, 3.6 Hz, 1H), 3.07–3.01 (m, 1H), 2.96–2.90 (m, 1H), 2.21–2.12 (m, 2H), 1.99–1.92 (m, 1H), 1.78–1.50 (m, 2H), 1.09 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.95, 143.52, 128.59, 127.18, 126.15, 60.58, 48.03, 47.24, 30.74, 26.16, 22.17; HRMS(ESI+) calcd for C13H19N2O [M + H]+ = 219.1497, found: 219.1498.
(S)-N-((S)-1-oxo-1-(((S)-1-phenylethyl)amino)propan-2-yl)pyrrolidine-2-carboxamide29f (14b)
Compound 14b was prepared from Boc-L-Pro-OH, Boc-L-Ala-OH and (S)-(−)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used.Yield 52%; white solid; m.p. 155–157 °C; [α]25D = −216.7° (c = 0.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.6 Hz, 1H), 7.37–7.26 (m, 5H), 6.96 (d, J = 7.2 Hz, 1H), 5.07 (t, J = 7.2 Hz, 1H), 4.44 (t, J = 7.2 Hz, 1H), 3.78–3.73 (m, 1H), 3.05–3.01 (m, 1H), 2.95–2.91 (m, 1H), 2.15–1.91 (m, 2H), 1.90 (br, 1H), 1.77–1.67 (m, 2H), 1.46 (d, J = 6.8 Hz, 3H), 1.36 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.62, 171.26, 143.34, 128.64, 127.23, 126.05, 60.39, 48.90, 48.50, 47.27, 30.69, 26.21, 22.13, 17.73; HRMS(ESI+) calcd for C16H24N3O2 [M + H]+ = 290.1869, found: 290.1871.
(S)-N-((S)-3-methyl-1-oxo-1-(((S)-1-phenylethyl)amino)butan-2-yl)pyrrolidine-2-carboxamide22a (14c, LLS)
Compound 14c was prepared from Boc-L-Pro-OH, Boc-L-Val-OH and (S)-(−)-1-phenylethylamine. Yield 59%; white solid; m.p. 141–142 °C; [α]25D = −98.2° (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 8.8 Hz, 1H), 7.34–7.26 (m, 5H), 6.76 (d, J = 7.6 Hz, 1H), 5.10 (t, J = 7.2 Hz, 1H), 4.18 (t, J = 8.2 Hz, 1H), 3.77 (dd, J = 4.8, 4.4 Hz, 1H), 3.08–3.02 (m, 1H), 2.97–2.91 (m, 1H), 2.19–2.10 (m, 3H), 1.97–1.89 (m, 1H), 1.76–1.69 (m, 2H), 1.47 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 175.36, 170.37, 143.26, 128.61, 127.27, 126.14, 60.48, 58.47, 48.92, 47.31, 30.98, 30.68, 26.16, 21.92, 19.48, 18.12; HRMS(ESI+) calcd for C18H28N3O2 [M + H]+ = 318.2182, found: 318.2188.(S)-N-((S)-3,3-dimethyl-1-oxo-1-(((S)-1-phenylethyl)amino) butan-2-yl)pyrrolidine-2-carboxamide (14d)
Compound 14d was prepared from Boc-L-Pro-OH, 3-Me-Boc-L-Val-OH and (S)-(−)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used. Yield 62%; white solid; m.p. 86–88 °C; [α]25D = −80.3° (c = 0.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.45 (d, J = 10.0 Hz, 1H), 7.37–7.26 (m, 5H), 6.94 (d, J = 5.6 Hz, 1H), 5.10 (t, J = 7.2 Hz, 1H), 4.31 (d, J = 10.0 Hz, 1H), 3.85 (br, 1H), 3.11–2.97 (m, 2H), 2.21–2.11 (m, 1H), 1.97–1.72 (m, 2H), 1.49–1.47 (m, 2H), 1.46 (d, J = 6.8 Hz, 3H), 0.93 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 174.43, 169.67, 143.43, 128.52, 127.19, 126.32, 60.48, 60.12, 58.40, 48.96, 47.20, 34.98, 30.96, 26.60, 26.04, 21.89, 18.44; HRMS(ESI+) calcd for C19H30N3O2 [M + H]+ = 332.2338, found: 332.2337.(S)-N-((S)-1-oxo-3-phenyl-1-(((S)-1-phenylethyl)amino) propan-2-yl)pyrrolidine-2-carboxamide (14e)
Compound 14e was prepared from Boc-L-Pro-OH, Boc-L-Phe-OH and (S)-(−)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used.Yield 53%; white solid, m.p. 107–109 °C; [α]25D = −15.4° (c = 0.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.8 Hz, 1H), 7.30–7.12 (m, 10H), 6.64 (d, J = 29.3, 7.5 Hz, 1H), 5.02 (t, J = 7.2 Hz, 1H), 4.60 (q, J = 8.0 Hz, 1H), 3.64 (dd, J = 4.0, 5.2 Hz, 1H), 3.20–2.77 (m, 4H), 2.07–2.02 (m, 2H), 1.75–1.68 (m, 1H), 1.64–1.49 (m, 2H), 1.36 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.71, 169.81, 143.26, 137.21, 129.28, 128.56, 127.17, 126.78, 126.02, 60.23, 58.42, 48.81, 47.09, 30.65, 25.92, 21.97, 18.44; HRMS(ESI+) calcd for C22H28N3O2 [M + H]+ = 366.2182, found: 366.2179.
(S)-N-((S)-3-methyl-1-oxo-1-(((R)-1-phenylethyl)amino)butan-2-yl)pyrrolidine-2-carboxamide (14f, LLR)
Compound 14f was prepared from Boc-L-Pro-OH, Boc-L-Val-OH and (R)-(+)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used.Yield 67%; white solid, m.p. 132–133 °C; [α]25D = −23.5° (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 9.2 Hz, 1H), 7.28–7.20 (m, 5H), 6.90 (d, J = 8.0 Hz, 1H), 5.11 (t, J = 7.2 Hz, 1H), 4.23 (dd, J = 7.6, 1.6 Hz, 1H), 3.59 (dd, J = 4.8, 4.4 Hz, 1H), 3.03–2.97 (m, 1H), 2.94–2.89 (m, 1H), 2.23–2.05 (m, 2H), 1.94–1.86 (m, 2H), 1.73–1.66 (m, 2H), 1.49 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 175.58, 170.28, 143.18, 128.58, 127.15, 126.11, 60.41, 58.22, 48.73, 47.26, 30.95, 30.68, 26.17, 22.07, 19.52, 18.23; HRMS(ESI+) calcd for C18H27N3O2 [M + H]+ = 318.2182, found: 318.2182.
(R)-N-((R)-3-methyl-1-oxo-1-(((S)-1-phenylethyl)amino)butan-2-yl)pyrrolidine-2-carboxamide (14g, DDS)
Compound 14g was prepared from Boc-D-Pro-OH, Boc-D-Val-OH and (S)-(−)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used.Yield 53%; white solid; m.p. 122–123 °C; [α]25D = +23.9° (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 9.2 Hz, 1H), 7.30–7.20 (m, 5H), 6.93 (d, J = 8.0 Hz, 1H), 5.11 (t, J = 7.2 Hz, 1H), 4.23 (dd, J = 7.6, 1.6 Hz, 1H), 3.61 (dd, J = 5.2, 4.0 Hz, 1H), 3.04–2.98 (m, 1H), 2.94–2.90 (m, 1H), 2.23–2.05 (m, 3H), 1.93–1.85 (m, 1H), 1.70 (t, J = 6.8 Hz, 2H), 1.50 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.38, 170.23, 143.15, 128.57, 127.16, 126.11, 60.37, 58.34, 48.79, 47.24, 30.74, 26.12, 22.07, 19.51, 18.24; HRMS(ESI+) calcd for C18H28N3O2 [M + H]+ = 318.2182, found: 318.2187.
(R)-N-((R)-3-methyl-1-oxo-1-(((R)-1-phenylethyl)amino)butan-2-yl)pyrrolidine-2-carboxamide (14h, DDR)
Compound 14h was prepared from Boc-D-Pro-OH, Boc-D-Val-OH and (R)-(+)-1-phenylethylamine. The same procedure as described above for the preparation of compound 14c was used. Yield 56%; white solid; m.p. 141–143 °C; [α]25D = +98.8° (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 9.2 Hz, 1H), 7.35–7.25 (m, 5H), 7.02 (d, J = 7.6 Hz, 1H), 5.10 (t, J = 7.2 Hz, 1H), 4.24 (dd, J = 7.6, 1.6 Hz, 1H), 3.74 (dd, J = 4.8, 4.4 Hz, 1H), 3.07–3.01 (m, 1H), 2.96–2.91 (m, 1H), 2.17–2.06 (m, 3H), 1.96–1.88 (m, 1H), 1.72 (t, J = 6.8 Hz, 2H), 1.46 (d, J = 7.2 Hz, 3H), 0.88 (d, J = 6.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 175.04, 170.42, 143.44, 128.85, 128.56, 127.19, 126.17, 60.46, 58.35, 48.92, 47.27, 30.98, 30.96, 26.08, 22.01, 19.44, 18.15; HRMS(ESI+) calcd for C18H28N3O2 [M + H]+ = 318.2182, found: 318.2187.(S)-N-((S)-1-(benzylamino)-3-methyl-1-oxobutan-2-yl)pyrrolidine-2-carboxamide (14i)
Compound 14i was prepared from Boc-L-Pro-OH, Boc-L-Val-OH and benzylamine. The same procedure as described above for the preparation of compound 14c was used.Yield 51%; white solid; m.p. 121–123 °C; [α]25D = −61.3° (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 9.2 Hz, 1H), 7.35–7.32 (m, 5H), 6.75 (s, 1H), 4.49 (dd, J = 9.2, 5.6 Hz, 1H), 4.40 (dd, J = 9.2, 5.6 Hz, 1H), 4.19 (t, J = 8.4 Hz, 1H), 3.68 (dd, J = 4.2, 4.0 Hz, 1H), 3.06–3.00 (m, 1H), 2.96–2.91 (m, 1H), 2.28–2.19 (m, 1H), 2.15–2.07 (m, 1H), 1.93–1.86 (m, 2H), 1.75–1.68 (m, 2H), 0.95 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 172.25, 171.14, 138.12, 128.64, 127.74, 127.41, 60.33, 58.46, 47.21, 43.44, 30.88, 30.52, 28.27, 19.52, 18.45; HRMS(ESI+) calcd for C17H26N3O2 [M + H]+ = 304.2025, found: 304.2029.
Typical procedure for direct aldol reactions by using dipeptide-like organocatalyst 14f in brine
To a mixed solvents of sat. aq. brine (0.5 mL) and ketone (0.5 mL) was added aldehyde (0.5 mmol), and the corresponding catalyst 14f (1.6 mg, 0.005 mmol). The reaction mixture was stirred at 25 °C for 24 h. The mixture was then quenched with a solution of saturated NH4Cl (0.5 mL), and extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4 and concentrated under reduced pressure to give a residue which was purified through a flash column chromatography with n-hexane/ethyl acetate (1:1, V/V) to afford the pure aldol adduct. All these aldol products are known compounds29,33,34 and the enantiomeric excess (ee) of the aldol adduct was determined by chiral HPLC analysis. For HPLC spectra of compounds 3a–j and 15a–k, please refer to ESI.†
Acknowledgements
We thank the National Science Foundation of China (no. 21372259 and 21172262) for financial support of this work.Notes and references
- (a) R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121 RSC; (b) M. Henrion, V. Ritleng and M. J. Chetcuti, ACS Catal., 2015, 5, 1283 CrossRef CAS.
- (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (b) S. Roscales and A. G. Csákÿ, Chem. Soc. Rev., 2014, 43, 8215 RSC; (c) V. Bisai, A. Bisai and V. K. Singh, Tetrahedron, 2012, 68, 4541 CrossRef CAS PubMed.
- (a) G. L. Beutner and S. E. Denmark, Angew. Chem., Int. Ed., 2013, 52, 9086 CrossRef CAS PubMed; (b) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (c) J.-F. Zhao, B.-H. Tan and T.-P. Loh, Chem. Sci., 2011, 2, 349 RSC.
- (a) B. List, Acc. Chem. Res., 2004, 37, 548 CrossRef CAS PubMed; (b) Q.-Y. Zhang, X.-L. Cui, L. Zhang, S.-Z. Luo, H. Wang and Y.-J. Wu, Angew. Chem., Int. Ed., 2015, 54, 5210 CrossRef CAS PubMed; (c) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS PubMed; (d) T. Miura, H. Kasuga, K. Imai, M. Ina, N. Tada, N. Imai and A. Itoh, Org. Biomol. Chem., 2012, 10, 2209 RSC.
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (b) B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS.
- (a) X.-H. Liu, L.-L. Lin and X.-M. Feng, Chem. Commun., 2009, 6145 RSC; (b) C. Palomo, M. Oiarbide and J. M. García, Chem. Soc. Rev., 2004, 33, 65 RSC; (c) K. Weidner, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2014, 53, 6150 CrossRef CAS PubMed; (d) W. Wan, W. Gao, G.-B. Ma, L. Ma, F. Wang, J. Wang, H.-Z. Jiang, S.-Z. Zhu and J. Hao, RSC Adv., 2014, 4, 26563 RSC; (e) A. A. Elmekawy, J. B. Sweeney and D. R. Brown, Catal. Sci. Technol., 2015, 5, 690 RSC; (f) S. Bhowmick, S. S. Kunte and K. C. Bhowmick, RSC Adv., 2014, 4, 24311 RSC.
- (a) C. Shen, F.-Y. Shen, G.-B. Zhou, H.-J. Xia, X.-Z. Chen, X.-G. Liu and P.-F. Zhang, Catal. Commun., 2014, 26, 6 CrossRef PubMed; (b) J. Agarwal and R. K. Peddinti, Eur. J. Org. Chem., 2012, 6390 CrossRef CAS PubMed.
- P. Revelou, C. G. Kokotos and P. Moutevelis-Minakakis, Tetrahedron, 2012, 68, 8732 CrossRef CAS PubMed.
- (a) S. Fotaras, C. G. Kokotos, E. Tsandi and G. Kokotos, Eur. J. Org. Chem., 2011, 1310 CrossRef CAS PubMed; (b) K. Arnold, A. S. Batsanov, B. Davies, C. Grosjean, T. Schütz, A. Whiting and K. Zawatzkya, Chem. Commun., 2008, 3879 RSC.
- A. S. Saiyed and A. V. Bedekar, Tetrahedron: Asymmetry, 2013, 24, 1035 CrossRef CAS PubMed.
- (a) A. J. A. Cobb, D. M. Shaw and S. V. Ley, Synlett, 2004, 558 CAS; (b) C. E. T. Mitchell, S. E. Brenner, J. García-Fortanet and S. Ley, Org. Biomol. Chem., 2006, 4, 2039 RSC; (c) L. Wynands, S. Delacroix, A. N. Van Nhien, E. Soriano, J. Marco-Contelles and D. Postel, Tetrahedron, 2013, 69, 4899 CrossRef CAS PubMed.
- T. Kano, O. Tokuda and K. Maruoka, Tetrahedron Lett., 2006, 47, 7423 CrossRef CAS PubMed.
- G. Guillena, M. del C. Hita and C. Nájera, Tetrahedron: Asymmetry, 2006, 17, 1493 CrossRef CAS PubMed.
- C. D. Maycock and M. R. Ventura, Tetrahedron: Asymmetry, 2012, 23, 1262 CrossRef CAS PubMed.
- L. Li, S.-H. Gou and F. Liu, Tetrahedron Lett., 2013, 54, 6358 CrossRef CAS PubMed.
- (a) P.-X. Zhou, S.-Z. Luo and J.-P. Cheng, Org. Biomol. Chem., 2011, 9, 1784 RSC; (b) J. Huang, G.-D. Chen, X.-K. Fu, C. Li, C.-L. Wu and Q. Miao, Catal. Sci. Technol., 2012, 2, 547 RSC; (c) L. Peng, L.-L. Wang, J.-F. Bai, L.-N. Jia, Y.-L. Guo, X.-Y. Luo, F.-Y. Wang, X.-Y. Xu and L.-X. Wang, Org. Biomol. Chem., 2011, 9, 4774 RSC; (d) A. Kumar, S. Singh, V. Kumar and S. S. Chimni, Org. Biomol. Chem., 2011, 9, 2731 RSC.
- T. He, K. Li, M.-Y. Wu, M.-B. Wu, N. Wang, L. Pu and X.-Q. Yu, Tetrahedron, 2013, 69, 5136 CrossRef CAS PubMed.
- S. Eymur, E. Akceylan, O. Sahin, A. Uyanik and M. Yilmaz, Tetrahedron, 2014, 70, 4471 CrossRef CAS PubMed.
- (a) J.-R. Chen, X.-L. An, X.-Y. Zhu, X.-F. Wang and W.-J. Xiao, J. Org. Chem., 2008, 73, 6006 CrossRef CAS PubMed; (b) B.-L. Zheng, Q.-Z. Liu, C.-S. Guo, X.-L. Wang and L. He, Org. Biomol. Chem., 2007, 5, 2913 RSC.
- For small peptides catalysed organic reactions, see: (a) G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722 RSC; (b) E. A. C. Davie, S. M. Mennen, Y.-J. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed; (c) M. Akiyama, K. Akagawa, H. Seino and K. Kudo, Chem. Commun., 2014, 50, 7893 RSC; (d) J. Duschmalé, S. Kohrt and H. Wennemers, Chem. Commun., 2014, 50, 8109 RSC; (e) A. F. de la Torre, D. G. Rivera, M. A. B. Ferreira, A. G. Corrêa and M. W. Paixão, J. Org. Chem., 2013, 78, 10221 CrossRef CAS PubMed.
- (a) C. E. Müller, D. Zell, R. Hrdina, R. C. Wende, L. Wanka, S. M. M. Schuler and P. R. Schreiner, J. Org. Chem., 2013, 78, 8465 CrossRef PubMed; (b) A. M. Vibhute, A. Vidyasagar, S. Sarala and K. M. Sureshan, Chem. Commun., 2012, 48, 2448 RSC; (c) M. Matsumoto, S. J. Lee, M. R. Gagné and M. L. Waters, Org. Biomol. Chem., 2014, 12, 8711 RSC.
- (a) D. K. Romney and S. J. Miller, Org. Lett., 2012, 14, 1138 CrossRef CAS PubMed; (b) G. Peris, C. E. Jakobsche and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 8710 CrossRef CAS PubMed.
- K. Akagawa, S. Takigawa, I. S. Nagamine, R. Umezawa and K. Kudo, Org. Lett., 2013, 15, 4964 CrossRef CAS PubMed.
- For organic reactions in aqueous media, see: (a) J. Mlynarski and S. Baś, Chem. Soc. Rev., 2014, 43, 577 RSC; (b) J. Huang, G.-D. Chen, X.-K. Fu, C. Li, C.-L. Wu and Q. Miao, Catal. Sci. Technol., 2012, 2, 547 RSC; (c) S. Bhowmick, S. S. Kunte and K. C. Bhowmick, RSC Adv., 2014, 4, 24311 RSC; (d) X.-L. Liu, B.-W. Pan, W.-H. Zhang, C. Yang, J. Yang, Y. Shi, T.-T. Feng, Y. Zhou and W.-C. Yuan, Org. Biomol. Chem., 2015, 13, 601 RSC; (e) S. Bhowmick and K. C. Bhowmick, Tetrahedron: Asymmetry, 2011, 22, 1945 CrossRef CAS PubMed.
- T. J. Dickerson and K. D. Janda, J. Am. Chem. Soc., 2002, 124, 3220 CrossRef CAS PubMed.
- A. Córdova, W. Notz and C. F. Barbas III, Chem. Commun., 2002, 3024 Search PubMed.
- Y. Hayashi, S. Aratake, T. Okano, J. Takahashi, T. Sumiya and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 5527 CrossRef CAS PubMed.
- V. Maya, M. Raj and V. K. Singh, Org. Lett., 2007, 9, 2593 CrossRef CAS PubMed.
- For small peptides catalysed aldol reactions, see: (a) S. Bayat, B. A. Tejo, E. Abdulmalek, A. B. Salleh, Y. M. Normia and M. B. A. Rahman, RSC Adv., 2014, 4, 38859 RSC; (b) P. Dziedzic, W.-B. Zou, J. Háfrenb and A. Córdova, Org. Biomol. Chem., 2006, 4, 38 RSC; (c) S. Bhowmick, S. S. Kunte and K. C. Bhowmick, RSC Adv., 2014, 4, 24311 RSC; (d) S. Fotaras, C. G. Kokotos and G. Kokotos, Org. Biomol. Chem., 2012, 10, 5613 RSC; (e) C. Berdugo, B. Escuder and J. F. Miravet, Org. Biomol. Chem., 2015, 13, 592 RSC; (f) F.-B. Chen, S. Huang, H. Zhang, F.-Y. Liu and Y.-G. Peng, Tetrahedron, 2008, 64, 9585 CrossRef CAS PubMed; (g) A. Psarra, C. G. Kokotos and P. Moutevelis-Minakakis, Tetrahedron, 2014, 70, 608 CrossRef CAS PubMed; (h) E. Machuca, Y. Rojas and E. Juaristi, Asian J. Org. Chem., 2015, 4, 46 CrossRef CAS PubMed; (i) P. Krattiger, R. Kovasy, J. D. Revell, S. Ivan and H. Wennemers, Org. Lett., 2005, 7, 1101 CrossRef CAS PubMed.
- (a) I. Triandafillidi, A. Bisticha, E. Voutyritsa, G. Galiatsatou and C. G. Kokotos, Tetrahedron, 2015, 71, 932 CrossRef CAS PubMed; (b) A. J. Pearson and S. Panda, Org. Lett., 2011, 13, 5548 CrossRef CAS PubMed; (c) A. Bisticha, I. Triandafillidi and C. G. Kokotos, Tetrahedron: Asymmetry, 2015, 26, 102 CrossRef CAS PubMed; (d) S. Chandrasekhar, K. Johny and C. R. Reddy, Tetrahedron: Asymmetry, 2009, 20, 1742 CrossRef CAS PubMed.
- For the increase of catalyst-loading leading to the decrease of enantioselectivity of the product in organocatalysis, by the reaction from kinetic to thermodynamic-control, see: (a) G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944 CrossRef CAS PubMed; (b) A. Kumar and S. S. Chimni, Tetrahedron, 2013, 69, 5197 CrossRef CAS PubMed; (c) By self-association phenomenon of the catalyst, see: H. B. Jang, H. S. Rho, J. S. Oh, E. H. Nam, S. E. Park, H. Y. Bae and C. E. Song, Org. Biomol. Chem., 2010, 8, 3918 RSC.
- (a) F. Kelleher, S. Kelly, J. Watts and V. McKee, Tetrahedron, 2010, 66, 3525 CrossRef CAS PubMed; (b) J.-G. Xin, L. Chang, Z.-R. Hou, D.-J. Shang, X.-H. Liu and X.-M. Feng, Chem.–Eur. J., 2008, 14, 3177 CrossRef CAS PubMed.
- X.-X. Song, A.-X. Liu, S.-S. Liu, W.-C. Gao, M.-C. Wang and J.-B. Chang, Tetrahedron, 2014, 70, 1464 CrossRef CAS PubMed.
- L. Wynands, S. Delacroix, A. N. Van Nhien, E. Soriano, J. Marco-Contelles and D. Postel, Tetrahedron, 2013, 69, 4899 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and HRMS of compounds 14a–i, HPLC charts of aldol products 3a–j and 15a–k can be found in the ESI. See DOI: 10.1039/c5ra07019h |
| This journal is © The Royal Society of Chemistry 2015 |