
Structure, morphology and catalytic properties of pure and alloyed Au–ZnO hierarchical nanostructures†
M. Cargnello‡
a,
D. Salaab,
C. Chenc,
M. D'Arienzob,
R. J. Gortec and
C. B. Murray*ad
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA, USA 19104. E-mail: cbmurray@sas.upenn.edu; Tel: +1-215-898-0588
bINSTM, Department of Materials Science, University of Milano-Bicocca, Via R. Cozzi 53, I-20125 Milano, Italy
cDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA, USA 19104
dDepartment of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA, USA 19104
First published on 5th May 2015
Abstract
A novel preparation of Au@ZnO and Au/Cd–ZnO structures is reported. The different morphologies of the two nanostructures are the result of either multiple or single nucleation events depending on the crystalline nature of the Au or Au/Cd seeds. Both samples are surprisingly active for CO oxidation and the water–gas shift-reaction (WGSR) despite the large size (6–8 nm) of the Au cores, and show interesting support effects when deposited on different oxides.
The preparation of colloidal heterostructures with controlled morphology is an important goal for several applications.1,2 In heterostructures, very often the sum is more than the parts. Electronic and geometric effects in morphologically controlled structures can indeed play an important role in determining the optical, electronic and catalytic properties of such systems. In catalysis, heterostructures with a core–shell configuration are particularly appealing not only because of the opportunity to tailor the metal–support interface, which sometimes defines the catalytic activity of the whole system, but also because the core–shell structure helps stabilize active phases against sintering, leaching and other deactivation phenomena.3,4
The other advantage of colloidal heterostructures is their solubility in organic solvents. For this reason, they can be treated like molecules and used and dispensed as required by the application. We showed in the past that this characteristic is especially useful to control the formation of active sites in solution, and then to transfer them onto a high-surface area support which makes the materials amenable for catalysis.5 Here, we describe a similar approach for Au@ZnO and Au/Cd–ZnO nanostructures.
The core–shell structures were prepared exploiting a seed-mediated approach. Compared to one-pot methods, our methodology allows the almost independent tailoring of the core and shell structures and results in a very high yield of the final colloidal heterostructures. Monodisperse Au nanocrystals (NCs) were prepared according to literature procedures6 and protected with 1-dodecanethiol. After being purified, they were re-dissolved in a solution containing the ZnO precursors, degassed and heated to 300 °C for 1 h. The final structures enclose a single Au NC at the core, surrounded by several single-crystalline ZnO NCs forming a shell resembling petals around a pistil (Fig. 1a–c).7 Remarkably, Au NCs maintain their pristine size and morphology despite the high temperatures of the synthesis, likely because of stabilization induced by ZnO precursors assembling around the metal surfaces. ZnO shows a wurtzite crystal phase, with lattice fringes and d-spacing (0.26 nm) clearly attributable to the (0002) planes of the wurtzite structure (Fig. 1c and d). This element also suggests that ZnO grows along the c-axis of the [0001] direction, following the anisotropic hexagonal crystal structure of wurtzite. The synthesis is highly reproducible (Fig. S1†).
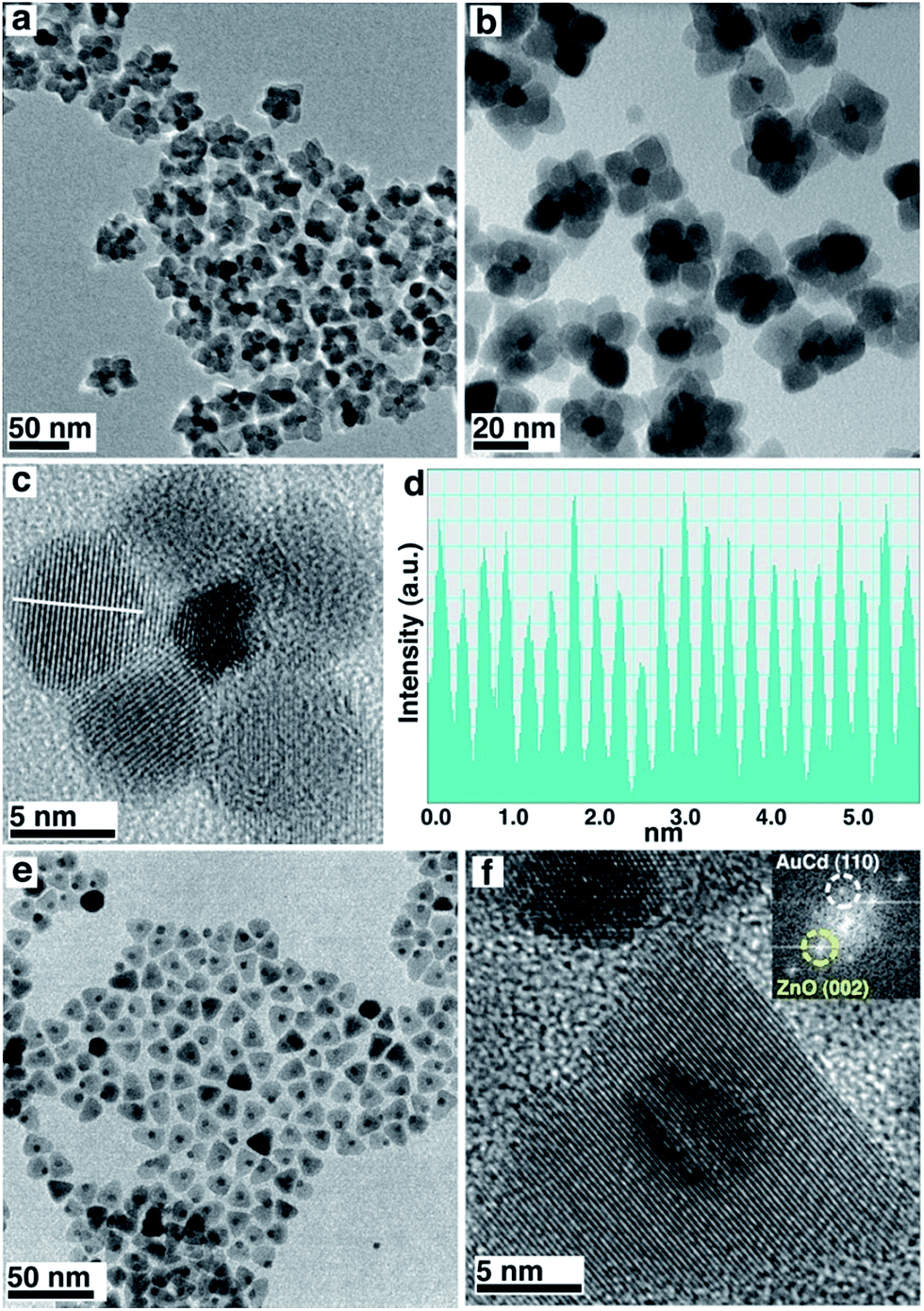 | ||
| Fig. 1 (a–c) TEM and HRTEM characterization of Au@ZnO core–shell structures (d) is the line-profile analysis shown in (c) demonstrating a lattice spacing of 0.26 nm. (e and f) TEM and HRTEM of Au/Cd–ZnO structures. Inset in (f) shows digital diffraction pattern of the nanostructures, highlighting the presence of the reflections attributable to AuCd (110) and ZnO (0002) as indicated by the circles. | ||
When the Au seeds are replaced by Au/Cd NCs, also prepared following literature procedures (as confirmed by XRD and HR-TEM, Fig. S2 and S3†),8 the morphology of the final structures changes (Fig. 1e and f). In this case, Au/Cd NCs are located at the tip of cone-like ZnO single crystals. The base of the cones is roughly hexagonal. This shape has been described in ZnO nanocrystals grown using a combination of an organic acid and an amine.9,10 Lattice fringes and d-spacing of the ZnO phase are still the same as for the bare Au case, demonstrating that Cd incorporation is likely limited to the Au seeds. TEM characterization and X-ray diffraction patterns of the two samples and of pure ZnO prepared with the same procedure, but with the exclusion of metallic seeds (Fig. S2†), showed that the Au/Cd–ZnO has indeed larger crystalline domains than Au@ZnO and bare ZnO, as evidenced by the sharper reflections in the XRD profile. This element confirms the single-crystalline nature of the ZnO structures bound to the Au and Au/Cd particles.
The hierarchical architecture of the final materials is dictated by the morphology of the starting Au seeds, and alloying Au with Cd promotes the formation of Au/Cd–ZnO structures with a distinct morphology compared to the bare Au structures. The different morphology and larger crystallite size in Au/Cd–ZnO suggests that the mechanism of ZnO formation follows different paths in the two samples. In particular, multiple vs. single nucleation events that differentiate the two structures must occur during ZnO formation (Fig. 2). We attribute the different mechanism to the structure of the seeds: Au NCs show multiple twin boundaries that are absent in the Au/Cd seeds, due to the high temperature annealing by which the latter are prepared (Fig. S3†). These defects may promote the multiple nucleation of ZnO onto the different Au facets, resulting in the core–shell type structure of the final materials. The Au/Cd seeds, instead, seem to favor the formation of only one ZnO nucleus on the particle surface, which then grows providing the final anisotropic structures (Fig. 2).11
 | ||
| Fig. 2 Scheme for the mechanism of formation of Au@ZnO from twinned Au seeds (top) and Au/Cd–ZnO structures from single-crystalline Au/Cd seeds (bottom). | ||
Both Au@ZnO and Au/Cd–ZnO colloidal structures were deposited onto three high-surface area supports, Al2O3, CeO2 and TiO2. The samples were calcined in air at 300 °C to fully remove the organic ligands and then tested for CO oxidation and water–gas shift-reaction (WGSR). It is well established that Au nanoparticles with size <5 nm generally show high catalytic activity because of electronic considerations,12 and that they are active WGS catalysts.13 More recently, support effects and metal-interface sites have also been indicated as active catalytic sites, providing another explanation for the special Au catalytic efficiency.14–16 In our case, the Au and Au/Cd particles in the final nanostructures were 6.1 and 8.2 nm in size, respectively, and therefore well above the dimensional limit considered for active catalysts. Nevertheless, the samples showed relevant activity for both investigated reactions, even if at high temperatures (Fig. 3). The bare supports did not show appreciable CO conversion under the conditions of our catalytic experiments. Even with the Au–ZnO interaction, especially in the Au@ZnO structures, a notable effect of the oxide support was found, with ceria-supported samples displaying higher rates than either titania- or alumina-supported ones. Furthermore, Au/Cd samples showed slightly improved activity compared to the bare Au ones, except for the ceria-supported systems, probably as a consequence of the formation of the Au2Cd intermetallic. Segregation of Cd and formation of CdO is likely to happen under CO oxidation and WGS conditions, and CdO may provide additional sites for activation of CO that are not present in the samples with bare Au nanoparticles. These sites improve the reactivity of the systems despite the larger metal particle size. In the case of support effects, a redox mechanism can be involved in the observed catalytic results. Ceria is known to provide activated oxygen species for both CO oxidation and WGSR.17,18 It is therefore possible that the Au particles, despite being in close contact with ZnO, may be affected by the presence of ceria, with the consequently higher rates for the reaction. This element would imply a long-range catalytic influence of ceria on Au. In the case of titania and alumina supports, the samples were much less active and required higher temperatures to provide detectable products. In any case, catalysts after reaction still showed the Au@ZnO structures to be intact (Fig. S4†), confirming the thermal stability of the systems.
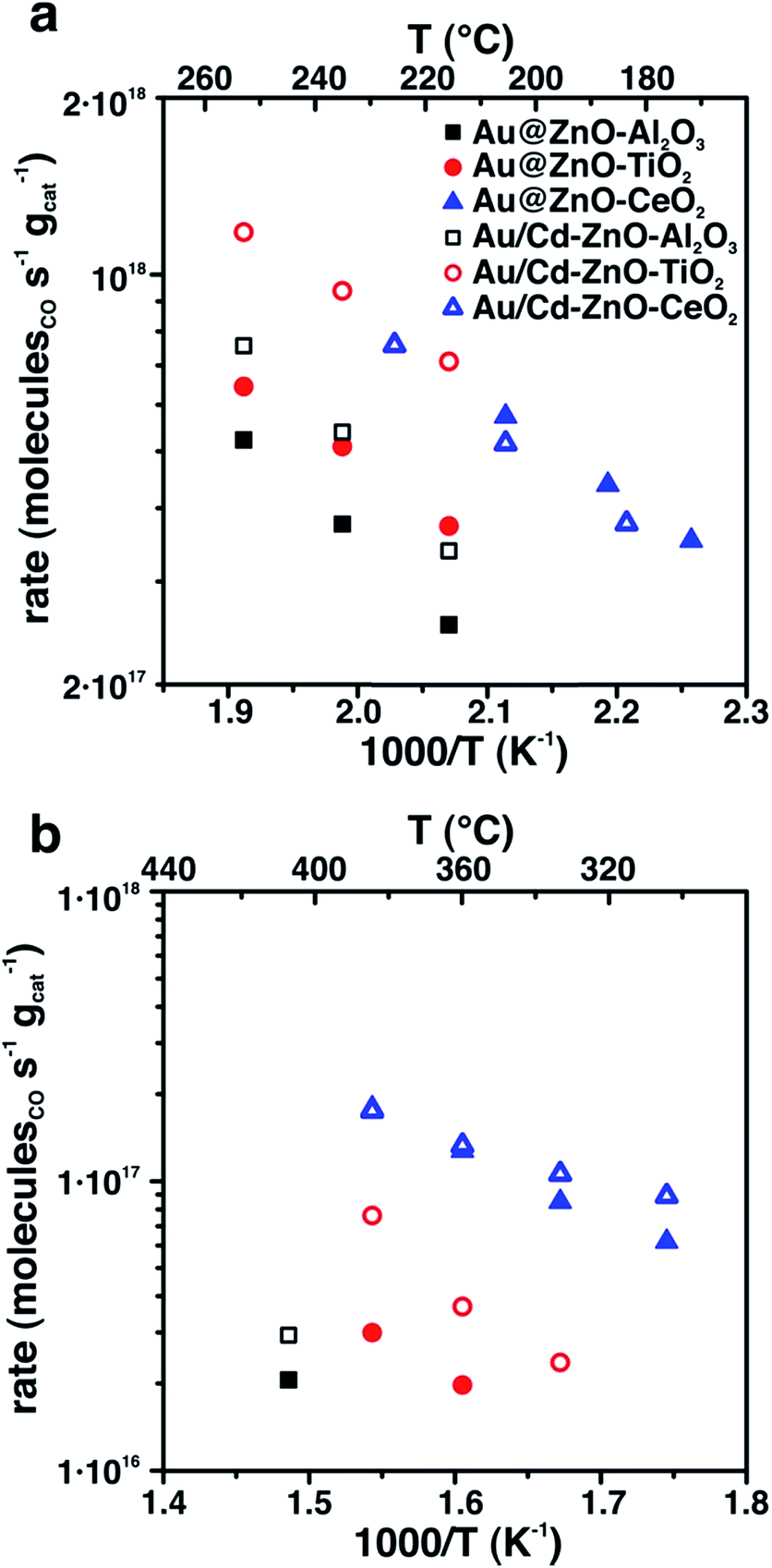 | ||
| Fig. 3 CO oxidation (a) and WGSR (b) activity of Au@ZnO and Au/Cd–ZnO structures supported on either alumina, titania or ceria. | ||
Conclusions
We reported the structural evolution of Au–ZnO nanostructures prepared by colloidal methods through a seed-mediated approach. Twin boundaries in Au seeds determine the formation of core–shell type structures, whereas by starting with Au/Cd seeds, a single crystal domain is obtained. Both structures are prepared with high morphological and dimensional control and, surprisingly, the metal nanoparticles do not change either their size or morphology during the synthesis. The structures show support effects for CO oxidation and WGSR.Acknowledgements
M.C. acknowledges support from the National Science Foundation through the Nano/Bio Interface Center at the University of Pennsylvania, Grant DMR08-32802. C.C. and R.J.G. were supported by the Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, Grant no. DE-FG02-13ER16380. C.B.M. is grateful for the support of the Richard Perry University Professorship. D.S and M.D gratefully acknowledge the financial support of EXTRA and EXCHANGE programs of the University of Milano-Bicocca.Notes and references
- L. Carbone and P. D. Cozzoli, Nano Today, 2010, 5, 449–493 CrossRef CAS PubMed
.
- M. Casavola, R. Buonsanti, G. Caputo and P. D. Cozzoli, Eur. J. Inorg. Chem., 2008, 2008, 837–854 CrossRef PubMed
- M. Cargnello, P. Fornasiero and R. J. Gorte, ChemPhysChem, 2013, 14, 3869–3877 CrossRef CAS PubMed
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed
- M. Cargnello, J. J. D. Jaén, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed
- S. Peng, Y. Lee, C. Wang, H. Yin, S. Dai and S. Sun, Nano Res., 2008, 1, 229–234 CrossRef CAS PubMed
- Y. Chen, D. Zeng, K. Zhang, A. Lu, L. Wang and D. L. Peng, Nanoscale, 2014, 6, 874–881 RSC
- P. Guardia, K. Korobchevskaya, A. Casu, A. Genovese, L. Manna and A. Comin, ACS Nano, 2013, 7, 1045–1053 CrossRef CAS PubMed
- J. Joo, S. G. Kwon, J. H. Yu and T. Hyeon, Adv. Mater., 2005, 17, 1873–1877 CrossRef CAS PubMed
- P. Li, Z. Wei, T. Wu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2011, 133, 5660–5663 CrossRef CAS PubMed
- N. P. Herring, K. AbouZeid, M. B. Mohamed, J. Pinsk and M. S. El-Shall, Langmuir, 2011, 27, 15146–15154 CrossRef CAS PubMed
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS
- Q. Fu, A. Weber and M. Flytzani-Stephanopoulos, Catal. Lett., 2001, 77, 87–95 CrossRef CAS
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed
- Y. Kang, X. Ye, J. Chen, L. Qi, R. E. Diaz, V. Doan-Nguyen, G. Xing, C. R. Kagan, J. Li, R. J. Gorte, E. A. Stach and C. B. Murray, J. Am. Chem. Soc., 2013, 135, 1499–1505 CrossRef CAS PubMed
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed
- J. Guzman, S. Carrettin, J. C. Fierro-Gonzalez, Y. Hao, B. C. Gates and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 4778–4781 CrossRef CAS PubMed
- T. Bunluesin, E. S. Putna and R. J. Gorte, Catal. Lett., 1996, 41, 1–5 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization techniques and supplementary figures. See DOI: 10.1039/c5ra06910f |
| ‡ Present address: Department of Chemical Engineering and SUNCAT Center for Interface Science and Catalysis, Stanford University, Stanford, CA 94305, USA. |
| This journal is © The Royal Society of Chemistry 2015 |