
Production of cellulose nanocrystals via a scalable mechanical method†
Abstract
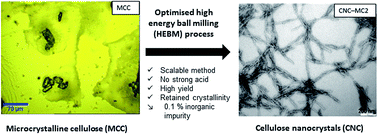
The production of rigid rod-like cellulose nanocrystals (CNC) via more scalable methods is necessitated by an increasing demand for CNC in various industrial sectors over the last few years. Contemporary protocols involve the consumption of large amounts of strong acids, enzymatic treatments, ultra-sonication and combinations thereof. In an attempt to address this scalability challenge, we aimed to isolate CNC via a scalable mechanical method i.e. high energy bead milling (HEBM). An aqueous dispersion of commercially available microcrystalline cellulose (MCC) was micronized through a HEBM process. This process was optimised by varying the concentration (0.5–2 wt%) and time (15–60 min) parameters, in order to obtain a high yield of well-separated CNCs as characterised by transmission electron microscopy (TEM). Micronisation of cellulose via the HEBM method under mild conditions resulted in cellulose nanocrystals with an average aspect ratio in the range of 20 to 26. The nanocrystals also retained both their crystallinity index (ICr) (85 to 95%) and thermal stability described in terms of onset degradation temperature (Tonset) (230–263 °C). The production yield of CNC from MCC via this process ranged between 57 and 76%. In addition, we found that micronisation of the MCC in the presence of dilute phosphoric acid also resulted in CNC with an average aspect ratio ranging from 21 to 33, high crystallinity (88–90%) and good thermal stability (Tonset 250 °C). In this study, we demonstrate the micronisation of commercially available MCC into CNC and describe their dimensions and properties after acid treatment and HEBM. Furthermore, we are able to recommend the use of this scalable milling process to produce rod-like cellulose nanocrystals having a thermal stability suitable to withstand the melt processing temperatures of most common thermoplastics.
 Please wait while we load your content...
Please wait while we load your content...

