
Bioavailability, plasma protein binding and metabolic stability studies of a ALDH2 activator, alda-1, using a validated LC-ESI-MS/MS method in rat plasma
Isha
Taneja†
ab,
Kanumuri Siva Rama
Raju†
ab,
Monika
Mittal
c,
Kapil
Dev
ad,
Mohammad Faheem
Khan
ad,
Rakesh
Maurya
ad and
Muhammad
Wahajuddin
*ab
aAcademy of Scientific and Innovative Research, New Delhi, India
bPharmacokinetics and Metabolism Division, CSIR- Central Drug Research Institute, Lucknow-226031, Uttar Pradesh, India. E-mail: wahajuddin@cdri.res.in; wahajuddin@gmail.com; Fax: +91-522-2771941; Tel: +91-522-2772450 ext. 4849 Tel: +91-522-2772550 ext. 4850
cDivision of Endocrinology, CSIR- Central Drug Research Institute, Lucknow, India
dMedicinal and Process Chemistry Division, CSIR- Central Drug Research Institute, Lucknow, India
First published on 22nd May 2015
Abstract
Alda-1 is an activator of the enzyme ALDH2. It has been suggested as a novel therapeutic for cardiovascular implications such as myocardial infarction, coronary bypass surgery, heart transplantation, peripheral artery disease, ischemia reperfusion injury, angina and alcoholic cardiomyopathy. Despite its widespread experimental use, no reports are available on its pharmacokinetics or bioanalytical quantification. In the present study, a simple, precise and reliable LC-ESI-MS/MS method has been developed and validated for the first time for quantification of alda-1 in plasma. Alda-1 was analyzed on a C18 column using methanol and 0.1% formic acid (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, v/v) as the mobile phase at a flow rate of 0.7 mL min−1. The method was found to be linear within the concentration range of 1–500 ng mL−1. The intra- and inter-day precision and accuracy were within acceptable limits. For the first time, the preclinical oral and intravenous pharmacokinetics of alda-1 were conducted. Alda-1 was found to be a rapidly absorbed, high clearance and poorly bioavailable compound in rats. Its plasma protein binding was found to be 82–86%. In view of the new regulatory guidelines, incurred sample reanalysis was also performed and all the samples were found within 15% of the mean value. From the in vitro microsomal incubation studies, it was found to be a high extraction compound. The data presented here provide important information to support the in vivo efficacy of alda-1 and would be helpful in its further development as a therapeutic agent and synthesis of its analogs with better systemic exposure and disposition properties.
:40, v/v) as the mobile phase at a flow rate of 0.7 mL min−1. The method was found to be linear within the concentration range of 1–500 ng mL−1. The intra- and inter-day precision and accuracy were within acceptable limits. For the first time, the preclinical oral and intravenous pharmacokinetics of alda-1 were conducted. Alda-1 was found to be a rapidly absorbed, high clearance and poorly bioavailable compound in rats. Its plasma protein binding was found to be 82–86%. In view of the new regulatory guidelines, incurred sample reanalysis was also performed and all the samples were found within 15% of the mean value. From the in vitro microsomal incubation studies, it was found to be a high extraction compound. The data presented here provide important information to support the in vivo efficacy of alda-1 and would be helpful in its further development as a therapeutic agent and synthesis of its analogs with better systemic exposure and disposition properties.
Introduction
Aldehyde dehydrogenases (ALDH) are a group of enzymes catalyzing NAD(P)+ dependent oxidation of aldehydes. The two most important isoforms responsible for converting aldehydes into non-toxic acids are its cytosolic form, ALDH1, and its mitochondrial form, ALDH2. The mitochondrial enzyme is the second enzyme in the oxidative detoxification of ethanol. It is also involved in the metabolism of toxic endogenous aldehydes such as 4-hydroxynonenal (4-HNE) and environmental aldehydic pollutants such as acrolein as well as in the bioconversion of nitroglycerin. Additionally, ALDH2 has also been implicated to play a cardio-protective role in ischemia induced heart damage during myocardial infarction and an inverse correlation has been suggested between ALDH2 activity and infarct size.1,2In approximately 40% of the East Asians, ALDH2 occurs as mutant ALDH2*2 due to a single nucleotide substitution of G to A in exon 12, resulting in glutamate at position 487 instead of lysine. This single amino acid polymorphism causes disruption of the co-enzyme NAD binding thereby leading to reduced catalytic activity. The mutant ALDH2*2 has 200 fold higher Km for NAD+ and 10 fold lower kcat as compared to the wild-type enzyme.3 As a result, the homozygous ALDH2*2 have 1–5% activity of the wild-type ALDH2*1 while the heterozygous ALDH2*1/*2 have 10–45% activity. In individuals having mutant ALDH2, acetaldehyde accumulates upon consumption of alcohol causing Asian flushing syndrome/alcohol flush reaction, characterized by facial flushing, nausea, drowsiness and increased heartbeat. The mutant ALDH2 also leads to decreased vasodilatory effect of nitroglycerin used in the treatment of angina, hypertension and myocardial infarction. An increased susceptibility to upper digestive tract cancers and neurodegenerative diseases has also been associated with ALDH2*2.
Alda-1 (Fig. 1), chemically N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide, is an activator of the enzyme ALDH2, acting as a structural chaperone and restoring the activity of ALDH2*2.1 It has been proposed that alda-1 promotes the apparent binding affinity for NAD+, the essential cofactor for the functioning of ALDH2. Thus, it reinstates the structural and functional conformation of the active site of ALDH2 resulting in its increased dehydrogenase, esterase and nitroglycerin denitration activity.4 The binding of alda-1 decreases the apparent Km for NAD+ by 6.7-fold and increases the Vmax by 2-fold.3 Due to the protective role of ALDH2 in regulation of free radical generation, metabolism of 4-HNE and mitochondrial dysfunction, activators of ALDH2 such as alda-1 that restore its activity have been advocated as novel therapeutics for cardiovascular implications such as myocardial infarction, coronary bypass surgery, heart transplantation, peripheral artery disease, ischemia reperfusion injury, angina and alcoholic cardiomyopathy.5,6 The in vivo cardio-protective role of alda-1 has been confirmed in myocardial infarction rat model. Administration of alda-1 at 8 mg kg−1 prior to ischemia reduced the infarct size by 60%.2
Despite the progressive research on its clinical utility and its popular use as an experimental tool, no literature is reported describing its pharmacokinetic and disposition aspects. Also, to the best of our knowledge, no method is reported for its quantification in biomatrix. Thus, in the present study, we have developed and validated an LC-ESI-MS/MS method for the quantification of alda-1 in rat plasma for the first time. We have also determined its in vivo preclinical pharmacokinetics, protein binding and in vitro metabolic profile for the first time. Understanding the pharmacokinetic and metabolic aspects of any therapeutic agent is important since its in vitro and in vivo efficacy is correlated to the systemic concentration and the exposure time. In vivo pharmacokinetic studies give an insight into the dose-exposure relationship while the in vitro metabolic profile provides useful information regarding elimination mechanism of the compound and rate of biotransformation, both of which decide the residence time of the compound in biological system. Since, it is only the free fraction of the drug that is available for exerting the pharmacodynamics response and that gets distributed and excreted from the body, the plasma protein binding studies becomes important. Thus, the data presented here provide important information to support the in vivo efficacy of alda-1 and would be helpful in its further development as a therapeutic agent and synthesis of its analogs with better systemic exposure and disposition properties.
Results and discussion
Optimization of LC-MS/MS conditions
Full quadrupole scans were carried out in positive ion mode to optimize ESI-MS/MS conditions. During the initial Q1 scan, the protonated molecular ions [M + H]+, [M + H + 2]+ and [M + H + 4]+ (m/z 323.9, 325.9 and 328.0) for alda-1 were observed in the ratio of 9:6:1 approximately, which is the typical mass spectra peak pattern for chlorine atom. Its sodium adduct was also observed in the similar isotopic pattern in the ratio of 9:6:1 for [M + Na]+, [M + Na + 2]+ and [M + Na + 4]+. For medicarpin (internal standard, IS), the protonated molecular ion [M + H]+ appeared at m/z 271.6. During the MRM scan, m/z 325.9 was found to have better signal-to-noise ratio than 323.9 or 328.0 and was chosen as the precursor ion for quantifying alda-1. Following detailed optimization of mass spectrometry conditions, the transitions m/z 325.9 precursor ion [M + H + 2]+ to the m/z 135.1 product ion and m/z 271.6 precursor ion [M + H]+ to the 137.1 m/z product ion were selected as the quantifier transition for alda-1 and IS, respectively. The transitions m/z 325.9 precursor ion [M + H + 2]+ to the m/z 77.2 product ion and m/z 325.9 precursor ion [M + H + 2]+ to the m/z 51 were selected as the qualifier transitions for alda-1. The ratio of the qualifier peak to the quantifier peak for m/z 77.2 product ion was found to be 0.175 and 0.170 for m/z product ion 55 and this ratio deviated within ±5.6% and ±5.5% for the respective transitions. The Q1 and product ion spectra of alda-1 and IS are shown in Fig. 2. The fragmentation pattern of alda-1 is shown in Fig. 3.
 | ||
| Fig. 1 Chemical structures of alda-1 and medicarpin (IS). | ||
 | ||
| Fig. 2 Q1 and product ion scans of (a) alda-1 and (b) IS. | ||
 | ||
| Fig. 3 Proposed fragmentation pattern of alda-1. | ||
The LC method was developed by examining the feasibility of various combinations of solvents such as acetonitrile and methanol as organic modifiers; and ammonium acetate, ammonium formate, formic acid and acetic acid as aqueous phase. Different C18 columns were tried along with altered mobile phase compositions and flow rates to achieve good peak shape, sensitivity and selectivity for alda-1 and IS. The LC method was optimized with mobile phase comprising of methanol and 0.1% formic acid in ratio of 60:40 (v/v) which was delivered at a flow rate of 0.7 mL min−1 for chromatographic separation on a Waters XBridge® C18 column (4.6 mm × 50 mm, 5.0 μm).
Method validation
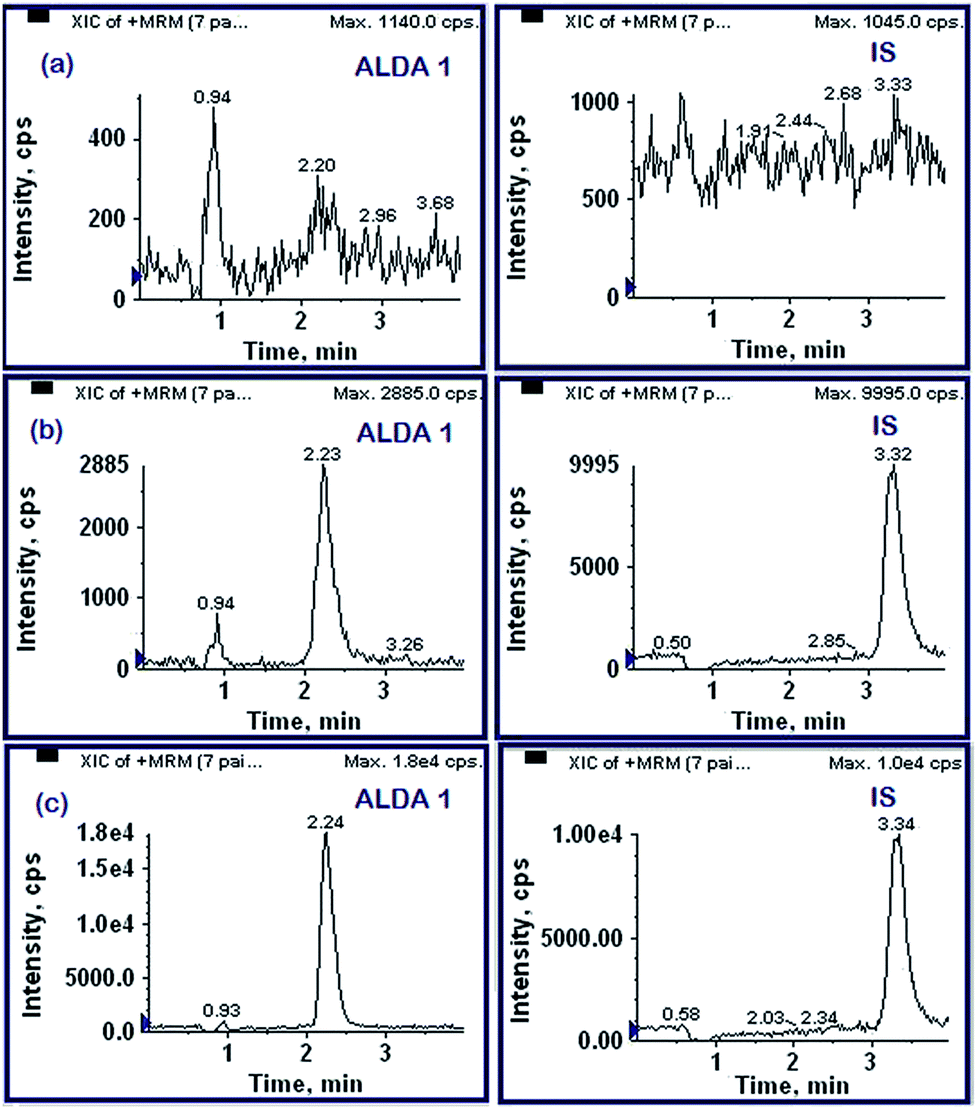 | ||
| Fig. 4 Typical MRM chromatograms of (a) drug free blank rat plasma, (b) drug free rat plasma spiked with alda-1 at LLOQ (1 ng mL−1) and IS and (c) 5 h in vivo study sample obtained upon intravenous administration. | ||
 | ||
| Fig. 5 Standard curve for alda-1 within the linearity range of 1–500 ng mL−1. | ||
| Alda-1 (ng mL−1) | ||||
|---|---|---|---|---|
| 1 (LLOQ) | 3 (LQC) | 150 (MQC) | 420 (HQC) | |
| a Calculated as (mean determined concentration/nominal concentration) × 100. b Expressed as %R.S.D. (S.D./mean) × 100. | ||||
| Intra-day | ||||
| Day 1 | ||||
| Mean ± SD | 0.91 ± 0.05 | 2.77 ± 0.11 | 156 ± 2.97 | 415.83 ± 20.36 |
| Accuracya (%) | 91.12 | 92.44 | 104.00 | 99.01 |
| Precisionb (%) | 5.14 | 4.06 | 1.90 | 4.90 |
|
||||
| Day 2 | ||||
| Mean ± SD | 1.06 ± 0.04 | 2.83 ± 0.25 | 142.83 ± 6.05 | 398.33 ± 9.44 |
| Accuracya (%) | 106.33 | 94.33 | 95.22 | 94.84 |
| Precisionb (%) | 3.29 | 8.67 | 4.23 | 2.37 |
|
||||
| Day 3 | ||||
| Mean ± SD | 1.01 ± 0.12 | 3.07 ± 0.36 | 140.17 ± 6.88 | 415.83 ± 20.36 |
| Accuracya (%) | 100.83 | 102.33 | 93.44 | 99.01 |
| Precisionb (%) | 11.78 | 11.63 | 4.91 | 4.90 |
|
||||
| Inter-day | ||||
| Mean ± SD | 0.95 ± 0.08 | 2.92 ± 0.29 | 152.67 ± 7.51 | 401.61 ± 22.60 |
| Accuracya (%) | 94.85 | 97.41 | 101.78 | 95.62 |
| Precisionb (%) | 8.00 | 9.82 | 4.92 | 5.63 |
| Alda-1 (ng mL−1) | ||||
|---|---|---|---|---|
| 1 (LLOQ) | 3 (LQC) | 150 (MQC) | 420 (HQC) | |
| Bench top stability (6 h) | ||||
| Mean ± SD | 1.00 ± 0.10 | 2.97 ± 0.19 | 150.33 ± 3.20 | 394.33 ± 19.99 |
| Accuracy | 99.16 | 96.85 | 96.37 | 94.83 |
| Precision | 10.27 | 6.52 | 2.13 | 5.07 |
|
||||
| Auto sampler stability (24 h) | ||||
| Mean ± SD | 0.99 ± 0.13 | 2.86 ± 0.18 | 155.50 ± 5.09 | 444.67 ± 22.92 |
| Accuracy | 98.20 | 93.00 | 99.68 | 106.93 |
| Precision | 13.51 | 6.17 | 3.27 | 5.16 |
|
||||
| Long term stability (30 days) | ||||
| Mean ± SD | 0.92 ± 0.06 | 2.92 ± 0.30 | 157.33 ± 6.57 | 381.17 ± 12.59 |
| Accuracy | 91.45 | 95.22 | 100.85 | 91.66 |
| Precision | 6.35 | 10.10 | 4.18 | 3.30 |
|
||||
| Freeze thaw stability (3 cycles) | ||||
| Mean ± SD | 1.02 ± 0.07 | 3.14 ± 0.25 | 138.50 ± 7.26 | 371.50 ± 8.69 |
| Accuracy | 101.47 | 102.28 | 88.78 | 89.34 |
| Precision | 7.07 | 8.06 | 5.24 | 2.34 |
In vitro plasma protein binding
The plasma protein binding studies were performed at two concentrations by filtration through anisotropic, hydrophilic YMT ultrafiltration membranes. One of the major drawbacks limiting the use of ultrafiltration technique for protein binding studies is the adsorption of the analyte on to filtration membrane. The non-specific binding studies were thus, carried out. The binding of alda-1 to YMT membrane was found to be insignificant (<10%), thus, validating the use of this technique for alda-1. The correction for non-specific binding was, however, taken into consideration. The mean plasma protein binding of alda-1 was found to be concentration independent (p > 0.05) and ranged between 82–86% at 100 and 1000 ng mL−1.In vitro metabolic stability and in vitro–in vivo extrapolation
The in vitro metabolic stability was performed to understand the biotransformation of alda-1 via cytochrome P450 (CYP450) pathway. Substrate depletion approach was used to estimate the in vitro t1/2, since the metabolites of alda-1 were unknown at this stage. The results are summarized in Table 3. The mean hepatic clearance in rats was found to be 53.49 mL min−1 kg−1 (1.28 L h−1 kg−1) and the mean hepatic extraction ratio was 0.76. The degradation profile of alda-1 in rat liver microsomes is given in Fig. 6. Around 10% of the parent remained in the microsomal reaction mixture after 60 minutes incubation.| Parameter | Alda-1 |
|---|---|
| Plasma protein binding (%) | 82.83 ± 1.81 (at 100 ng mL−1) |
| 85.94 ± 2.15 (at 1000 ng mL−1) | |
| In vitro t 1/2 (min) | 11.01 ± 0.93 |
| Intrinsic clearance (CLint) (μL min−1 kg−1) | 126.3 ± 10.63 |
| Whole liver clearance (mL min−1 kg−1) | 227.34 ± 19.14 |
| Hepatic clearance (mL min−1 kg−1) | 53.49 ± 1.06 |
| Hepatic extraction ratio | 0.76 ± 0.01 |
 | ||
| Fig. 6 Degradation profile of alda-1 upon incubation with rat liver microsomes (mean ± S.D). | ||
In vivo pharmacokinetics
The developed method was used to determine the pharmacokinetics of alda-1 in female SD rats after oral and intravenous administration. The data was subjected to non-compartmental analysis. The mean plasma concentration–time profiles are presented in Fig. 7 and the pharmacokinetic parameters in Table 4. | ||
| Fig. 7 Mean plasma concentration–time plots of alda-1 following intravenous (10 mg kg−1) and oral (40 mg kg−1) administration (mean ± S.D). | ||
| Parameter | Intravenous (10 mg kg−1) | Oral (40 mg kg−1) |
|---|---|---|
| t 1/2 (h) | 1.67 ± 0.54 | 7.33 ± 1.37 |
| C o (ng mL−1) | 8774.71 ± 955.72 | — |
| C max (ng mL−1) | — | 69.88 ± 4.60 |
| T max (h) | — | 0.63 ± 0.25 |
| V d (L kg−1) | 9.39 ± 1.80 | 41.50 ± 2.95 |
| CL (L h−1 kg−1) | 3.97 ± 0.54 | 4.01 ± 0.72 |
| AUC0–t (h × ng mL−1) | 2536.07 ± 347.25 | 415.83 ± 57.04 |
| AUC0–∞ (h × ng mL−1) | 2540.80 ± 246.19 | 469.12 ± 81.24 |
| Bioavailability (%) | — | 4.62 |
Upon oral administration, the peak concentration was reached at 0.63 h and was found to be 69.88 ng mL−1. This suggests that alda-1 has a high absorption rate. It was found to have a low oral bioavailability of 4.62%. Upon intravenous administration, alda-1 was detectable up to 9 h only. The high clearance, 3.97 ± 0.54 L h−1 kg−1, of alda-1 suggest it to be a high extraction compound. However, the in vivo clearance was found to be significantly higher than the hepatic clearance (1.28 L h−1 kg−1) obtained from microsomal studies. This implies that apart from CYP-mediated phase I biotransformation, other elimination mechanisms such as phase II biotransformation or renal elimination might also be involved that are responsible for the high in vivo clearance of this compound. The high hepatic extraction of alda-1 and its poor bioavailability could prove to be a limiting factor in clinical development of this compound. During the reanalysis of the incurred samples, all the samples were found to be within ±15% of the mean value, thus, re-establishing the validity and robustness of the developed method.
Materials and methods
Chemicals and reagents
Alda-1 was provided by Dr. Naibedya Chattopadhyay from Division of Endocrinology, CSIR-Central Drug Research Institute (Lucknow, India). Medicarpin (internal standard, IS) was synthesized at the Medicinal Chemistry Division of Central Drug Research Institute (Lucknow, India). HPLC grade methanol, acetonitrile and sodium carboxy methyl cellulose (CMC) were purchased from Sigma Aldrich Ltd (St Louis, USA). Formic acid AR was purchased from E Merck Limited (Mumbai, India). Centrifree® micro partition system (product no. 4104) was purchased from Amicon Inc. (MA, USA). Ultra pure water was obtained from a Sartorious Arium 611 system. Heparin sodium injection I.P. (1000 IU per mL, Biologicals E. Limited, Hyderabad, India) was purchased from local pharmacy.Animals and prerequisites: blank, drug free plasma samples were collected from adult, healthy female Sprague–Dawley (SD) rats at the Division of Laboratory Animals (DOLA) of Central Drug Research Institute (Lucknow, India). Prior approval from the Institutional Animal Ethics Committee (IAEC) was sought for maintenance, experimental studies, euthanasia and disposal of carcass of animals. Plasma was obtained by centrifuging the heparinised blood (25 IU per mL) at 2000× g for 10 min at 20 °C.
Instrumentation and LC-ESI-MS/MS conditions
Chromatographic separation was performed on a Waters XBridge® C18 column (4.6 × 50 mm, 5.0 μm) using Shimadzu UFLC consisting of SCL 20Avp pump and SIL-HTc autosampler. The system was run in isocratic mode with mobile phase consisting of methanol:0.1% formic acid in the ratio of 60:40 (v/v) at a flow rate of 0.7 mL min−1. Mobile phase was duly filtered through 0.22 μm Millipore filter (Billerica, USA) and degassed ultrasonically for 15 min prior to use. Separations were performed at room temperature. The injection volume was kept at 20 μL.
Mass spectrometric detection was performed in multiple reaction monitoring (MRM) mode on an API 4000 QTRAP mass spectrometer (Applied Biosystems, MDS Sciex Toronto, Canada) equipped with an API electrospray ionization (ESI) source and triple quadrupole mass analyser. Quadrupoles Q1 and Q3 were set on unit resolution in positive ion mode. The ion spray voltage was set at 5500 V. The instrument parameters viz., nebulizer gas, curtain gas, auxillary gas and collision gas were set at 50, 20, 50 and 8 psi, respectively. Compound related parameters viz., declustering potential (DP), collision energy (CE), entrance potential (EP) and collision cell exit potential (CXP) were 66, 29, 10, 26 V; and 50, 25, 10, 10 V for alda-1 and IS, respectively. Zero air was used as source gas while nitrogen was used as both curtain and collision gas. The transition of m/z 325.9 precursor ion [M + H + 2]+ to the m/z 135.1 product ion was monitored for alda-1 and m/z 271.6 precursor ion [M + H]+ to the 137.1 m/z product ion for IS. Data acquisition and quantitation were performed using Analyst® software version 1.6 (Applied Biosystems, MDS Sciex Toronto, Canada).
Stocks and working standards
A primary stock solutions of alda-1 and IS (1 mg mL−1) were prepared in methanol. Working standard solutions were prepared by combining the aliquots of each primary stock solution and diluting with methanol. All the solutions were stored at −20 °C. The stability of the stock solutions were confirmed by comparing them with the freshly prepared stock solutions.Calibration standards and quality control samples
Calibration standards of alda-1 were prepared by spiking 5 μL of appropriate working stocks in 95 μL pooled blank rat plasma to obtain nine calibration standards from 1–500 ng mL−1. Calibration curves were obtained by plotting the peak area ratios of the analyte to IS versus the analyte concentration. The curves were fitted using linear regression with 1/x2 weighting. The quality control (QC) samples were prepared at four concentrations levels, 1 ng mL−1 (lower limit of quantitation, LLOQ), 3 ng mL−1 (low QC), 150 ng mL−1 (medium QC) and 420 ng mL−1 (high QC), by following the same procedure as for calibration standards. The lowest limit of detection (LOD) was defined as the lowest concentration with signal-to-noise ratio more than 3. The LLOQ was defined as the lowest concentration with signal-to-noise ratio more than 10 and having accuracy and precision within ±20%. All samples were stored at −70 °C until analysis.Sample preparation
A simple protein precipitation technique was employed for extracting alda-1 from rat plasma. To 100 μL of rat plasma, 200 μL of acetonitrile (containing IS, equivalent to 50 ng mL−1) was added. The mixture was vortexed for 5 minutes followed by centrifugation for 10 min at 13000 g on Sigma 3-16K (Frankfurt, Germany). The supernatant was separated and 20 μL was injected for LC-MS/MS analysis.
Validation
The assay was validated following the bioanalytical method validation guidelines issued by the FDA Center for Drug Evaluation and Research (CDER).7 The parameters recovery, specificity, accuracy, precision, matrix effects, dilutional integrity and stability were evaluated using drug-free plasma from six different rats at four QC levels (LLOQ, QC low, QC medium and QC high). Incurred sample reanalysis was also performed to increase the reliability of the developed method.Specificity was tested by analyzing plasma samples spiked with alda-1 and IS in comparison to six randomly selected blank plasma samples to investigate potential interferences at the LC peak region of both the analytes. Recovery was assessed by comparing the peak responses of the pre-extracted plasma samples to those of post-extracted blank plasma samples spiked with equivalent concentration of the analytes. The recovery of IS was determined at 50 ng mL−1. The matrix effect was evaluated by comparing the peak response of the post extracted spiked plasma samples and the unextracted analytical neat samples at equivalent concentrations.8,9 The intraday accuracy and precision were determined by six repeat analyses (intrabatch variability) of the four QC concentrations. The interday accuracy and precision were determined by analyzing four QC concentrations in three different runs (interbatch variability) on 3 days. Accuracy should lie within ±15% deviation of the nominal value and precision within ±15% relative standard deviation (RSD) within except for LLOQ where both should lie within ±20%. The stability of plasma samples was assessed under different storage conditions: bench top (BT) stability at room temperature for 6 h; long term (LT) stability at −80 °C for 30 days, autosampler (AS) stability at 4 °C for 24 h and; freeze/thaw (FT) stability after 3 freeze/thaw cycles. Autosampler carry over was determined in triplicate by injecting the highest calibration standard followed by a blank sample and was considered insignificant if the measured peak area was less than 20% of the lowest calibrator area. Dilution integrity was performed by 20 times dilution of the plasma samples containing 8000 ng mL−1 of alda-1 with blank plasma to obtain plasma samples containing 400 ng mL−1 of alda-1. The response after dilution should not vary more than 15% of the true value. An incurred sample reanalysis (ISR) was performed by selecting the in vivo pharmacokinetic study samples, to show reproducibility of method. Four samples were selected from each rat including the time points at Cmax and elimination phase to cover the entire profile of the individual subject. Thus, the ISR was performed on 24 samples out of total 88 samples. The difference between the original and reanalyzed concentrations of the analyte should be within 20% for at least 67% of the total samples reanalyzed.7

In vitro plasma protein binding
The plasma protein binding of alda-1 was estimated in triplicate by ultra-filtration method using centrifree® micro partition system (Amicon Inc., MA, USA). Freshly collected rat plasma was spiked with alda-1 (stock solutions were prepared as described above) to obtain the plasma concentrations of 100 and 1000 ng mL−1. Spiked plasma was allowed to equilibrate for 15 min before the start of the study. Samples in triplicate were placed in centrifree® tubes and centrifuged at 3500× g for 15 min at 37 °C. 100 μl each of initial plasma samples and ultra-filtrates were subjected to LC-MS/MS after sample treatment as described previously.10 The non-specific binding (NBS) to the filtration membrane was estimated by spiked equivalent concentration in phosphate buffer saline (PBS) and further processing the samples similar to the plasma samples. The plasma protein binding was determined by using the formula:where CO is the dosing concentration of the compound; Cf is the concentration in the filtered matrix and; NBS is the non-specific binding calculated as 1 − (Cp,f/Cp,uf) where Cp,f is the average concentration in the filtered PBS and Cp,uf is the average concentration in the unfiltered PBS. Student's t-test of significance was applied to compare the effect of concentration on plasma protein binding of alda-1.

In vitro metabolic stability
Metabolic stability of alda-1 was performed in triplicate using glass tubes.11 To each tube, 460 μL of phosphate buffer (0.1 M, pH 7.4) and 12.5 μL of rat liver microsomal protein (20 mg mL−1) were added and incubated for at 37 ± 0.2 °C for 5 min. Then, 2.5 μL of test compound (1 mM) was added. For positive control, testosterone was used as the test compound. In negative control, NADPH was replaced by 25 μL of phosphate buffer. Reaction was initiated by addition of 25 μL of NADPH (24 mM) and terminated at 0, 5, 10, 15, 30, 45 and 60 min by addition of 450 μL ice-cold acetonitrile to 50 μL reaction mixture, followed by centrifugation for 15 min at 10000 rpm. 20 μL of supernatant was directly analyzed by LC-MS/MS. The data was analysed using GraphPad Prism 5.0 software. Non-linear regression of percent parent remaining against time was performed as per the equation:| %Parent drug remaining = span × e−kt + plateau |
The in vitro parameters were estimated using the following formulae:



where SF is the scaling factor defining microsomal protein per gram liver and was taken as 45 for rats. Q is the blood flow in rats taken as 70 mL min−1 kg−1.

In vivo pharmacokinetic study
Female SD rats were divided into two groups (n = 4 each): one group (for intravenous pharmacokinetic study) was provided free access to food and water; while the other group was fasted overnight (12–14 h) prior to the oral pharmacokinetic study and had free access to water throughout the experimental period. Alda-1 was administered orally at a dose of 40 mg kg−1 in 0.25% CMC suspension and intravenously as a solution (prepared in 40% dimethyl formamide, 30% polyethylene glycol, 20% triple distilled water and 10% ethanol) at a dose of 10 mg kg−1. Animals were provided with standard diet 3 h after oral dosing. The rats were partially anaesthetized using diethyl ether and blood samples were collected from the retro-orbital plexus into heparinized microfuge tubes at 0.083, 0.25, 0.5, 1, 1.5, 2, 3, 5, 7, 9 and 24 h post-dosing. Plasma was harvested by centrifuging the blood at 13000 rpm for 10 min on Sigma 1-15K (Frankfurt, Germany). Samples were stored at −80 ± 10 °C until bioanalysis. All the four QC concentrations (n = 5 each) were analysed with each batch of study samples. The observed maximum serum concentration (Cmax) and the time to reach the maximum serum concentration (Tmax) were obtained by visual inspection of the experimental data. The data was subjected to non-compartmental pharmacokinetic analysis using WinNonlin (version 5.1, Pharsight Corporation, Mountain View, USA). Area under curve (AUC) from 0 to 24 h (AUC0–24) was calculated using linear trapezoidal rule. AUC from 0 to infinity (AUC0–∞) was calculated as the sum of AUC0–t and Clast/kel, where, Clast represents the last quantifiable concentration and kel represents the elimination rate constant.
Conflict of interest
The authors declare that they have no conflict of interest.Conclusion
In the present study, an accurate, sensitive and robust LC-ESI-MS/MS method was developed and validated to quantify alda-1 in plasma biomatrix for the first time. The sensitivity of the method was 1 ng mL−1. The method was successfully applied to study the oral and intravenous pharmacokinetics alda-1. Alda-1 was found to be poorly bioavailable. The same method was also used to study its in vitro CYP stability and plasma protein binding. Alda-1 was found to be a high extraction compound and was 82–85% plasma protein bound. The study provides groundwork to carry forward the research in this direction.Acknowledgements
The authors are thankful to Director, CDRI for his constant encouragement and support. We also acknowledge Council of Scientific and Industrial Research (CSIR) for providing research fellowship to Isha Taneja and Kanumuri Siva Rama Raju. CDRI communication number is 9002.References
- C. H. Chen, G. R. Budas, E. N. Churchill, M. H. Disatnik, T. D. Hurley and D. Mochly-Rosen, Science, 2008, 321, 1493–1495 CrossRef CAS PubMed.
- G. R. Budas, M. H. Disatnik and D. Mochly-Rosen, Trends Cardiovasc. Med., 2009, 19, 158–164 CrossRef CAS PubMed.
- S. Perez-Miller, H. Younus, R. Vanam, C. H. Chen, D. Mochly-Rosen and T. D. Hurley, Nat. Struct. Mol. Biol., 2010, 17, 159–164 CAS.
- M. Beretta, A. C. Gorren, M. V. Wenzl, R. Weis, M. Russwurm, D. Koesling, K. Schmidt and B. Mayer, J. Biol. Chem., 2010, 285, 943–952 CrossRef CAS PubMed.
- C. H. Chen, L. Sun and D. Mochly-Rosen, Cardiovasc. Res., 2010, 88, 51–57 CrossRef CAS PubMed.
- A. Sun, Y. Zou, P. Wang, D. Xu, H. Gong, S. Wang, Y. Qin, P. Zhang, Y. Chen, M. Harada, T. Isse, T. Kawamoto, H. Fan, P. Yang, H. Akazawa, T. Nagai, H. Takano, P. Ping, I. Komuro and J. Ge, J. Am. Heart Assoc., 2014, 3, e000779 CrossRef PubMed.
- Guidance for Industy : Bioanalytical Method Validation, US Food and Drug Administration, 2013 Search PubMed.
- S. P. Singh, Wahajuddin, M. M. Ali, K. Kohli and G. K. Jain, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 2845–2851 CrossRef CAS PubMed.
- Wahajuddin, S. P. Singh and G. K. Jain, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1133–1139 CrossRef CAS PubMed.
- S. P. Singh, Wahajuddin, D. Tewari, T. Pradhan and G. K. Jain, Food Chem. Toxicol., 2011, 49, 1056–1062 CrossRef CAS PubMed.
- P. Baranczewski, A. Stanczak, K. Sundberg, R. Svensson, A. Wallin, J. Jansson, P. Garberg and H. Postlind, Pharmacol. Rep., 2006, 58, 453–472 CAS.
Footnote |
| † Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2015 |