
Aluminum interaction with 2,3-diphosphoglyceric acid. A computational study†
Abstract
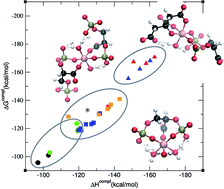
The interaction of aluminum with 2,3-diphosphoglyceric acid (2,3-DPG) is thought to be one of the strongest interactions of aluminium with a biophosphate molecule. In this article, the affinity energies for a family of Al–(2,3-DPG) complexes are calculated at the DFT level of theory. The study includes a total of 26 structures that vary from 1 : 1 complexes, to 1 : 2 stoichiometry and ternary complexes with citrate, considering different coordination modes and protonation states. Our results demonstrate that in the case of 1 : 1 complexes, the 2,3-DPG ligand could compete with citrate for complexation with aluminum at physiological pH. However, for the rest of the complexes there is a clear preference for Al(Citr)2 > Al(2,3-DPG)(Citr) ternary complex > Al(2,3-DPG)2 complexes. For each of these groups the charge of the ligand determines the affinity but to a lower extent than the nature of the Citr/2,3-DPG ligands. In summary, our results point to a high variety of possible complexation modes of 2,3-DPG to aluminum, showing a higher preference towards the formation of ternary complexes.
 Please wait while we load your content...
Please wait while we load your content...

