
Intermolecular OHN hydrogen bond with a proton moving in 3-methylpyridinium 2,6-dichloro-4-nitrophenolate†
Irena Majerz*a and
Matthias J. Gutmannb
aFaculty of Pharmacy, Wroclaw Medical University, Borowska 211a, 50-556 Wroclaw, Poland. E-mail: majerz@yahoo.com; irena.majerz@umed.wroc.pl
bRutherford Appleton Laboratory, ISIS Facility, Chilton Didcot, Oxfordshire OX11 0QX, UK
First published on 2nd November 2015
Abstract
The neutron structure of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate measured at different temperatures is used to discuss the structural parameters of the OHN intermolecular hydrogen bridges with the proton moving. The experimental and optimized structures, with the precise location of the proton, are used to explain the mechanism of proton transfer in the OHN hydrogen bridge.
Introduction
The importance of the hydrogen bond is well known, as it is very ubiquitous in biological systems which are responsible for the molecular and macroscopic properties of materials, molecular recognition, and supramolecular structure.1 The most important problem in investigating the hydrogen bond is the degree of proton transfer in the hydrogen bridge, which determines all the properties of the hydrogen bond. In molecular complexes the proton is located at the donor atom and in ionic pair-type complexes it shifts to the acceptor, but the most interesting are the strongest hydrogen bonds, with the proton located near the hydrogen bond middle, for which the degree of proton transfer is about 50%. The parameters which determine the location and movement of the proton in the hydrogen bond have been intensively investigated for many years.2 The main parameter influencing the location of the proton in the intermolecular hydrogen bond is the affinity of the proton donor and proton acceptor.3 Another parameter is the value ΔpKa (ΔpKa = pKb base − pKa acid).4 At low values of ΔpKa the proton is connected with the donor atom and at high ΔpKa it is shifted to the proton acceptor, so the degree of proton transfer depends on the substituents of proton donor and proton acceptor, which determine the ΔpKa value. For a narrow range of ΔpKa values, the proton can be located neither at the donor nor at the acceptor, but near the center of the hydrogen bridge. These very strong hydrogen bonds are, in addition to the properties of the proton donor and acceptor, also sensitive to temperature and the surroundings of the hydrogen bridge.5Among the intermolecular hydrogen bonds, the most typical are the complexes of phenols with tertiary amines. They form single hydrogen bonds without chains or bifurcated hydrogen bonds with strengths covering the whole range of hydrogen bonds, from molecular to very strong bonds with the proton at the center of the hydrogen bridge, up to ionic complexes with the proton shifted to the acceptor nitrogen atom. The complexes of phenols with tertiary amines are used as model complexes to investigate the interatomic OHN hydrogen bond. Throughout the long period of their investigation, many correlations linking the degree of proton transfer with structural,6 physicochemical,7 and spectroscopic parameters8 have been published. The most essential step in investigating the hydrogen bond is to determine its structure, with the precise location of the proton engaged in the hydrogen bond.
The structural parameters which can characterize the degree of proton transfer in the OHN hydrogen bond in phenol–tertiary amine complexes are the bond lengths connected with the hydrogen bridge, i.e. O⋯N, OH, and NH. The most characteristic is the O⋯N distance. For the strongest hydrogen bonds it is very short, below 2.6 Å. The proton in strong hydrogen bonds is located in the middle of the bridge, so the OH and NH distances are similar. Elongation of O⋯N is connected with a shifting of the proton to the donor or to the acceptor molecule, according to the properties of the donor and acceptor in the hydrogen bond complex. The OHN angle exhibits a general tendency to be linear when the O⋯N bridge is shortest. Some distances of the phenol molecule are also sensitive to the proton transfer, so the C–O distance can be used as a measure of the degree of proton transfer.
The mutual dependencies of O⋯N with OH and NH shown in9 have parabolic shape, and the correlation between OH and NH is sigmoidal. All these distances can be correlated with the value ΔpKa (ΔpKa = pKb base − pKa acid).
The dependencies of the structural parameters of the hydrogen bridge obtained experimentally for phenol–amine complexes have been compared with these parameters predicted using theoretical methods.10 Experimental structural parameters follow those calculated at the B3LYP/6-311++G** level. In this situation, one may expect that on the basis of the ΔpKa value of the phenol–amine complex the degree of proton transfer in the OHN bond can be predicted as well as all the structural parameters sensitive to the location of the proton in the hydrogen bridge.
Among numerous complexes with intermolecular hydrogen bond only few are characterized by very short distance between the proton donor and proton acceptor. Neutron structures of these complexes with the O⋯N distances shorter than 2.6 Å measured at different temperatures confirmed possibility of the proton moving along the hydrogen bridge upon the temperature.11,12
In this paper, the neutron structure of the of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate that is a typical phenol–pyridine complex is shown. The ΔpKa value of this complex equals 1.54, suggesting a short O⋯N distance and with possible symmetric location of the proton in the hydrogen bridge. Previously investigated X-ray structure showed that the parameters of the OHN hydrogen bond (O⋯ − 2.544(4), OH – 1.60(3), NH – 0.97(3), CO – 1.285(4) Å, OHN – 165(1)° (ref. 13)) are typical for very short hydrogen bond in which the proton can move along the hydrogen bridge upon the temperature change. The location of the hydrogen bonded proton by X-ray diffraction has very low precision, and therefore to investigate the details of the proton moving the neutron structure of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate has been measured. The aim of this study is a precise determination of the structural parameters of the hydrogen bond in the investigated complex at different temperature. The short hydrogen bond has been characterized by calculation of the potential energy curves for the proton moving in the hydrogen bridge. Combination of the experimental neutron structures and theoretical structures optimized for different location of the proton in the hydrogen bridge gives possibility to follow the proton transfer mechanism investigated by NBO analysis.
Experimental
2,6-Dichloro-4-nitrophenol was obtained by nitration of 2,6-dichlorophenol (Aldrich). The complexes were obtained by crystallization of 2,6-dichloro-4-nitrophenol with an excess of 3-methylpyridine from an acetone solution. The crystals were grown by dissolving the complex in CCl4 and slowly evaporating of the solvent.Neutron diffraction data in the temperature range from 10 K to 290 K were collected using the SXD instrument at the ISIS facility at Rutherford Appleton Laboratory (UK).14 To facilitate efficient data collection, two crystals were comounted and exposed simultaneously in the neutron beam, similar as was done in.15 Data were treated using the locally available SXD2001 software16 and refinements carried out using Jana2006.17 Details of the data collection and refinement parameters are summarized in Table 1.
| Diffractometer | SXD neutron time-of-flight Laue diffractometer | ||||||||||
| Wavelength (Å) | 0.37–8.8 | ||||||||||
| Compound | 3-Methylpyridinium 2,6-dichloro-4-nitrophenolate, C12H10N2O3Cl2 | ||||||||||
| Molecular weight | 301.1 | ||||||||||
| Unit cell | Monoclinic, space group P21/c, Z = 4 | ||||||||||
| T (K) | 10 | 30 | 50 | 80 | 110 | 140 | 170 | 200 | 230 | 260 | 290 |
| a (Å) | 7.7629(16) | 7.762(2) | 7.766(2) | 7.780(2) | 7.792(2) | 7.808(2) | 7.827(2) | 7.846(2) | 7.865(2) | 7.889(2) | 7.911(2) |
| b (Å) | 23.525(5) | 23.519(7) | 23.537(7) | 23.541(7) | 23.565(7) | 23.582(7) | 23.619(7) | 23.647(7) | 23.681(8) | 23.720(8) | 23.760(8) |
| c (Å) | 7.3900(18) | 7.387(2) | 7.395(2) | 7.405(2) | 7.419(2) | 7.432(2) | 7.447(2) | 7.460(2) | 7.472(3) | 7.487(3) | 7.502(3) |
| β (°) | 111.49(2) | 111.47(2) | 111.51(2) | 111.59(2) | 111.70(2) | 111.81(2) | 111.88(2) | 112.01(2) | 112.10(2) | 112.22(3) | 112.33(2) |
| V (Å3) | 1255.8(5) | 1255.0(7) | 1257.6(7) | 1261.1(7) | 1265.8(7) | 1270.4(6) | 1277.6(7) | 1283.2(7) | 1289.6(7) | 1297.0(8) | 1304.4(7) |
| ρcalc (g cm−3) | 1.59 | 1.59 | 1.59 | 1.59 | 1.59 | 1.57 | 1.57 | 1.56 | 1.55 | 1.54 | 1.53 |
| Number of refls | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 207 207 |
9693 | 9008 | 7985 | 7478 | 6697 | 5943 | 5593 | 4943 | 4651 | 4056 |
| Unique refls I > 2σ | 9069 | 8591 | 7930 | 6946 | 6546 | 5808 | 5219 | 4973 | 4370 | 4120 | 3638 |
| N parameters | 268 | ||||||||||
| R(F) | 0.0937 | 0.0991 | 0.0901 | 0.0794 | 0.0891 | 0.0962 | 0.0884 | 0.0861 | 0.0850 | 0.0883 | 0.0817 |
| wR(F2) | 0.1775 | 0.1871 | 0.1601 | 0.1603 | 0.1638 | 0.1849 | 0.1649 | 0.1591 | 0.1606 | 0.1708 | 0.1525 |
| Extinction coeff. | 33.8(7) | 35.1(8) | 36.3(7) | 37.2(8) | 35.5(9) | 40.9(9) | 45.0(10) | 48.3(12) | 46.7(13) | 50.6(14) | 53.4(15) |
| Absorption coeff. | 2.770 + 0.0155λ | ||||||||||
Ab initio calculations based on the Born–Oppenheimer approximation were carried out at the B3LYP/6-311++G** level using the Gaussian 09 program.18 For the experimental structures the proton in the hydrogen bond was moved with the step of 0.05 Å, within the range 0.8–2 Å to calculate the potential energy surfaces. The vibrational time-independent Schrödinger equation for the OH coordinate was solved numerically using the two-boundary-conditions approach basing on the Numerov algorithm19 with the related variational-theory-based program.20 The structure optimized when the OH distance was kept constant and the experimental structures were used to perform the Natural Bond Orbital (NBO) analysis using NBO5.0 program.21 The wave function evaluated for each molecule was used as the input to the AIMALL program.22
Results and discussion
1. Structure of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate
The molecular structure of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate determined by neutron diffraction is shown in Fig. 1 and the structural parameters of the OHN hydrogen bond at different temperatures are collected in Table 2.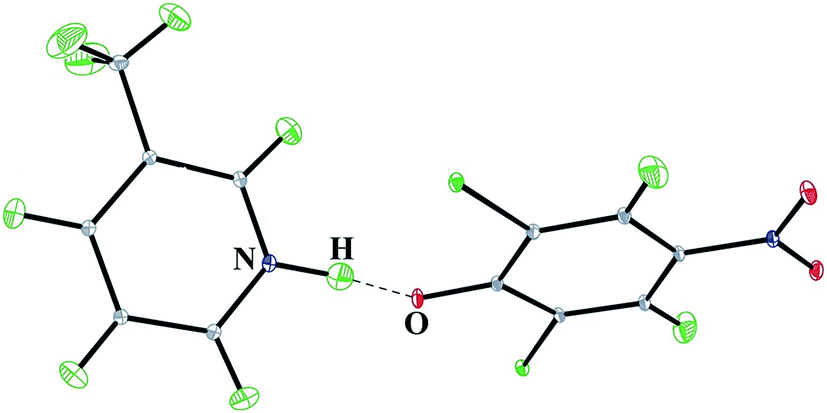 | ||
| Fig. 1 Crystal structure of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate from single crystal neutron diffraction. Thermal displacement ellipsoids are drawn at the 50% probability level. | ||
| T [K] | N⋯O [Å] | OH [Å] | NH [Å] | CO [Å] | OHN [°] |
|---|---|---|---|---|---|
| 290 | 2.533(5) | 1.386(8) | 1.167(8) | 1.271(6) | 167.0(7) |
| 270 | 2.532(4) | 1.400(8) | 1.152(8) | 1.270(5) | 165.8(7) |
| 230 | 2.532(4) | 1.398(8) | 1.151(8) | 1.272(5) | 166.8(7) |
| 200 | 2.531(4) | 1.406(8) | 1.141(7) | 1.269(4) | 167.3(7) |
| 170 | 2.531(3) | 1.405(6) | 1.143(5) | 1.275(3) | 166.8(5) |
| 140 | 2.531(3) | 1.412(5) | 1.137(5) | 1.271(3) | 166.3(5) |
| 110 | 2.523(3) | 1.436(5) | 1.116(5) | 1.281(4) | 162.7(5) |
| 80 | 2.531(3) | 1.418(5) | 1.131(5) | 1.277(3) | 166.3(5) |
| 50 | 2.537(2) | 1.429(4) | 1.126(4) | 1.275(2) | 166.3(4) |
| 30 | 2.5416(6) | 1.4210(3) | 1.1363(3) | 1.2759(4) | 167.24(2) |
| 10 | 2.5340(4) | 1.4264(2) | 1.1294(2) | 1.2726(3) | 164.93(2) |
According to the ΔpKa value of this complex, that equals 1.54, the OHN hydrogen bond should be very short, below 2.6 Å. Indeed, the O⋯N bond length in 3-methylpyridinium 2,6-dichloro-4-nitrophenol measured at different temperatures changes from 2.5416(6) to 2.523(3) Å. Very short distance between the proton donor and proton acceptor is usually connected with location of the proton in the middle of the distance between the proton donor and proton acceptor. Despite this expectation, the proton in the OHN hydrogen bond in 3-methylpyridinium 2,6-dichloro-4-nitrophenolate is shifted to the acceptor. Temperature increasing moves the proton to the central location in the hydrogen bond. Probably at higher temperatures the OH and NH distances can be equalized. Previously the X-ray structure of the complex of 2,6-dichloro-4-nitrophenol with 3-methylpyridine was investigated,13 and the N⋯O, OH, NH, and CO bond lengths were 2.544(4), 1.60(3), 0.93(3), and 1.285(4) Å respectively. Very short N⋯O bond length should be connected with location of the proton in the middle of the O⋯N distance, but both: neutron and X-ray structural measurements locate the proton at the acceptor. Neutron diffraction allows a precise determination of the proton location in the hydrogen bridge. X-ray diffraction measurement of the structure of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate showed that the proton is shifted to the acceptor. Precisely determined proton position in the neutron measurement shows that the OH and NH distances are more equalized and the difference between OH and NH is less significant than the determined in X-ray diffraction. Nonsymmetrical location of the proton can be explained by special properties of the strong hydrogen bond. Unlike to molecular and ionic hydrogen bonds, the strong hydrogen bond is sensitive to temperature, environment, and isotopic substitution. Neutron diffraction confirms sensitivity of the proton location to temperature. Probably electric properties of the crystal of the investigated compound are responsible for shifting of the proton to the acceptor atom.
The cell parameters in Table 1 undergo systematic changes with temperature (a = 1E-06T2 + 0.0002T + 7.7559, R2 = 0.9972, b = 2E-06T2 + 0.0002T + 23.518, R2 = 0.9958, c = 4E-07T2 + 0.0003T + 7.3809, R2 = 0.9942, β = 3E-06T2 + 0.0021T + 111.42, R2 = 0.9938) so a phase transition is excluded. The distances connected with the OHN hydrogen bond listed in Table 2 change not continuously. Limited temperature changes of the N⋯O distance are close to 3σ value. Changes of NH and OH are more significant and linear correlation between NH and OH (NH = −0.9007OH + 2.411, R2 = 0.9488) reflects the proton moving in the hydrogen bond caused by temperature.
Correlations linking the geometrical hydrogen bond parameters have been investigated for very long time.6,23 Fig. 2 collects correlations for the neutron structures of O⋯N hydrogen bond complexes with the proton moving upon temperature compared with theoretical curves.24 The experimental data from the neutron structures with precisely determined proton position reproduces and confirms theoretical correlations linking the parameters of the hydrogen bridge: the Bond Order Reaction Curve (BORC) curve25 in Fig. 2a and the theoretical curve for the hydrogen bond length and deviation of the proton from central location between the proton donor and acceptor – Fig. 2b. When the experimental data confirm the theoretical correlation linking OH and NH, the theoretical curve in Fig. 2b calculated without taking into account anharmonicity of the hydrogen bridge vibrations, can be compared with the correlation obtained with the experimental neutron data. Anharmonicity correction can be neglected for molecular and ionic OHN hydrogen bonds but is significant for the strongest hydrogen bonds. Except anharmonicity correction also the quantum nature of the proton in short hydrogen bond influences the O⋯N distance and it can be expected that for the investigated complex this effect should be about −0.02 Å.26 Experimental neutron data for the complexes with proton moving in the hydrogen bridge allow reproducing the shape of the experimental curve and deliver the anharmonicity corrections.
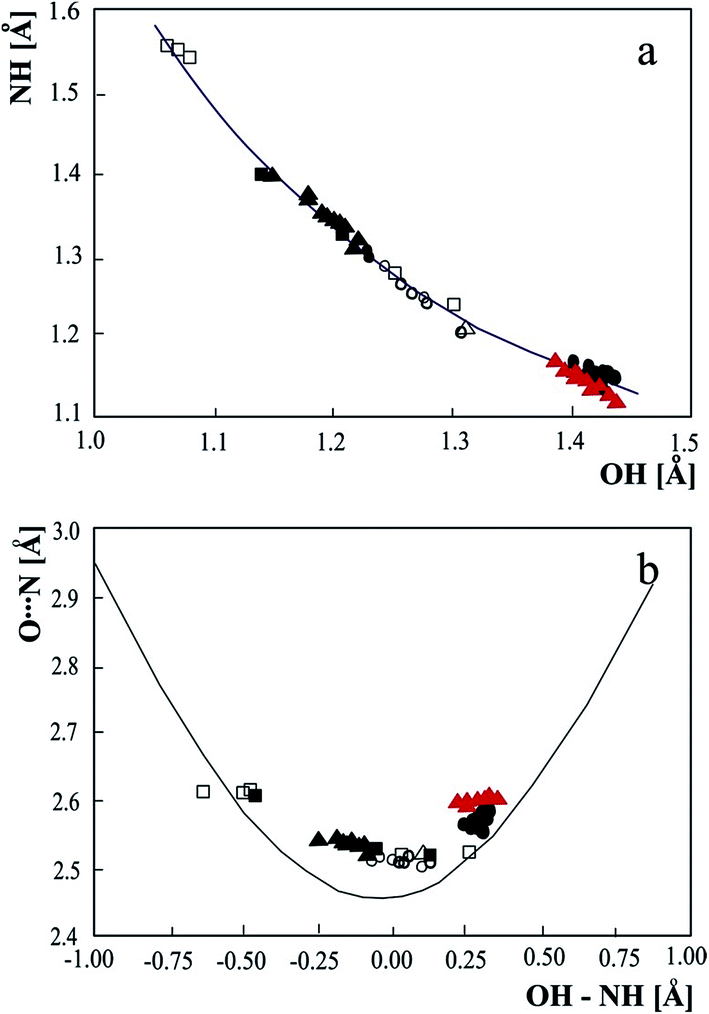 | ||
| Fig. 2 Correlations of the geometric parameter of the strong OHN hydrogen bonds with proton moving in the hydrogen bridge: (a) – relation between NH and OH bond lengths, (b) – dependence of N⋯O bridge length as a function of the difference between OH and NH. The experimental neutron bridge parameters are compared with the theoretical curve calculated according to BORC curve (a) and p1 = exp(−(r1 − r1o)/b1), p2 = exp(−(r2 − r2o)/b2), p1 + p2 = 0, r1 = rOH, r2 = rNH, where p1 and p2 are the corresponding valence bond orders of the diatomic units, b1 = 0.371, r1o = 0.942, b2 = 0.385, r2o = 0.992 Å (b). Experimental data: • – pyridinium 2,4-dinitrobenzoate,27 o – 4-methylpyridine – pentachlorophenol complex,28 □ – 1:2 adduct of benzene-1,2,4,5-tetracarboxylic acid and 4,4′-bipyridyl,29 Δ – pyridine-3,5-dicarboxylic acid,30 ▲ – complex of 3,5-dinitrobenzoic acid with 3,5-dimethylpyridine,12 ▲ (red) – 3-methylpyridinium 2,6-dichloro-4-nitrophenolate. | ||
The CO bond length of the phenolic part of the complex, used as a measure of the degree of proton transfer in the intermolecular hydrogen bond, changes from 1.34 Å in molecular complexes to 1.27 Å in ionic complexes. For very short hydrogen bonds the CO bond length should be between these two values, although it has been shown that CO bond length as a function of OH depends on the substituents of the phenol ring.10 In 3-methylpyridinium 2,6-dichloro-4-nitrophenolate the CO bond length found in neutron measurement is typical of ionic hydrogen-bonded complexes, which is in agreement with the location of the proton at the acceptor when this same length for the X-ray structure equals 1.285(4) Å. The CO and OH bond lengths of the 2,6-dichloro-4-nitrophenol – 3-methylpyridine complex do not fulfill the general sigmoidal correlation10,31 linking the OH and CO bond lengths.
2. Potential energy dependencies for proton moving in 3-methylpyridinium 2,6-dichloro-4-nitrophenolate
To illustrate sensitivity of the proton to temperature, the potential energy curve for moving the proton along the O⋯N bridge has been calculated for the 2,6-dichloro-4-nitrophenol – 3-methylpyridine complex with both geometries: this obtained in the neutron at 290 K and in the X-ray measurements.13 Both curves are shown in Fig. 3.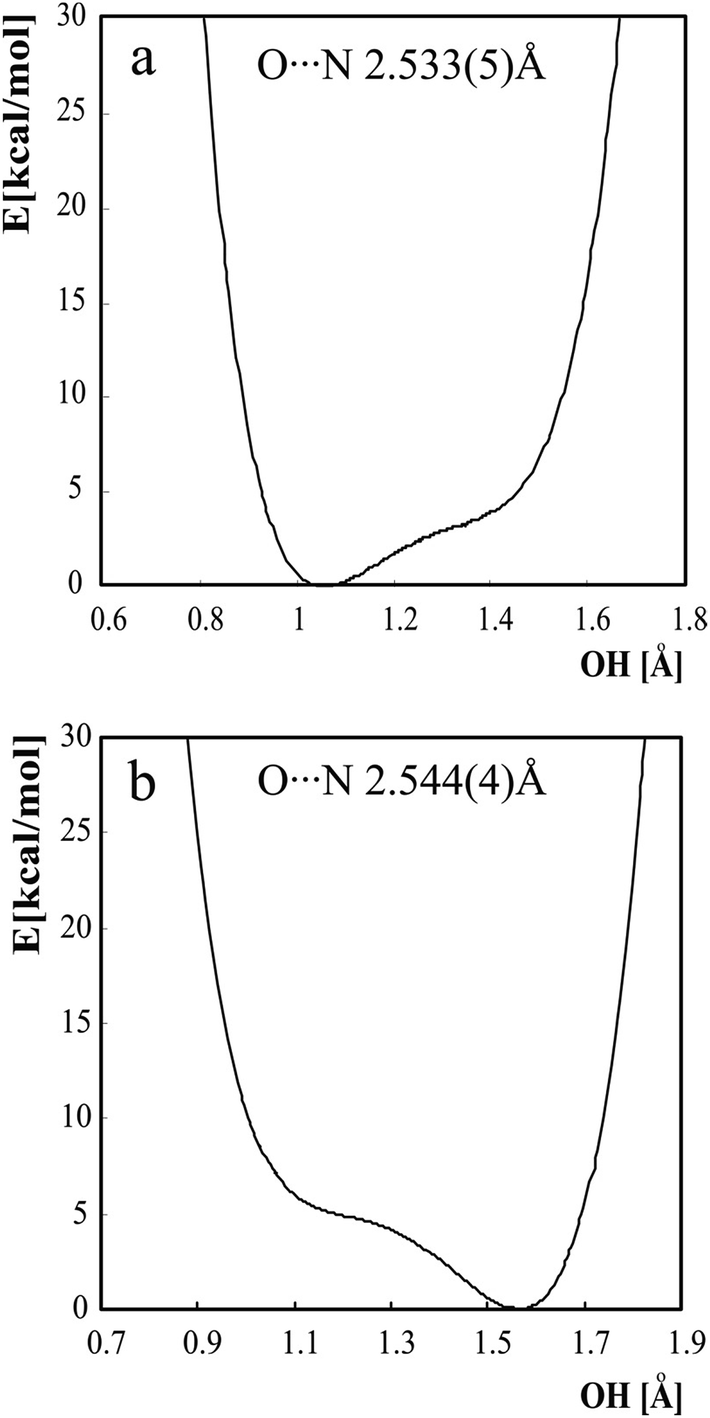 | ||
| Fig. 3 Potential energy curves for moving a proton in the OHN hydrogen bond in the 2,6-dichloro-4-nitrophenol with 3-methylpyridine complex. (a) – curve calculated moving the proton along the O⋯N bond of length equal to 2.533(5) Å as in the neutron structure at 290 K, (b) – this same curve performed using the O⋯N distance of 2.544(4) Å as in the X-ray structure. | ||
Both curves are characteristic of a strong hydrogen bond. A very broad minimum spreads over a broad range of OH. The lowest energy value is located at an OH distance which shifts the proton to the acceptor as in the X-ray structure or to proton donor as in the neutron structure. The change in the O⋯N bond length from 2.544(4) to 2.533(5) Å causes a significant difference in the OH distance, characteristic for the energy minimum. The shape of the curve does not make a determination of the height of the energy barrier between the two minimums possible because the second minimum is not well shaped. It is characteristic that the small difference in the geometry of the molecule with regard to the neutron and X-ray structures causes a shift of the minimum of the potential energy curve for proton movement in the hydrogen bridge. For the X-ray structure (O⋯N = 2.544(4) Å) the minimum on the potential energy curve is located at the OH bond length of 1.56 Å for and for the neutron structure (O⋯N = 2.533(5) Å) the minimum is shifted to the OH bond length of 1.05 Å. In both curves, the first energy level is located at higher energy compared with the top of the barrier on the potential energy curve, so the proton can move easily along the hydrogen bridge and it is very easy to shift it from the donor to the acceptor atom. For the curves in Fig. 3 the highest frequencies of vibrational transitions are equal 4523 cm−1 for the curves calculated for O⋯N distance characteristic for the neutron structure and 3975 cm−1 for the distance characteristic for the X-ray structure. Analysis of the vibrational properties of strong hydrogen bond should involve tunneling and quantum nuclear effects32 but even rough analysis exhibits difference between both vibrational transitions and emphasizes sensitivity of the experimental properties to the O⋯N distance.
To investigate the energy as a function of two parameters of the hydrogen bridge, the potential energy surfaces presented in Fig. 4 have been calculated. The potential energy surfaces have been calculated for relaxed geometry except the fixed distances.
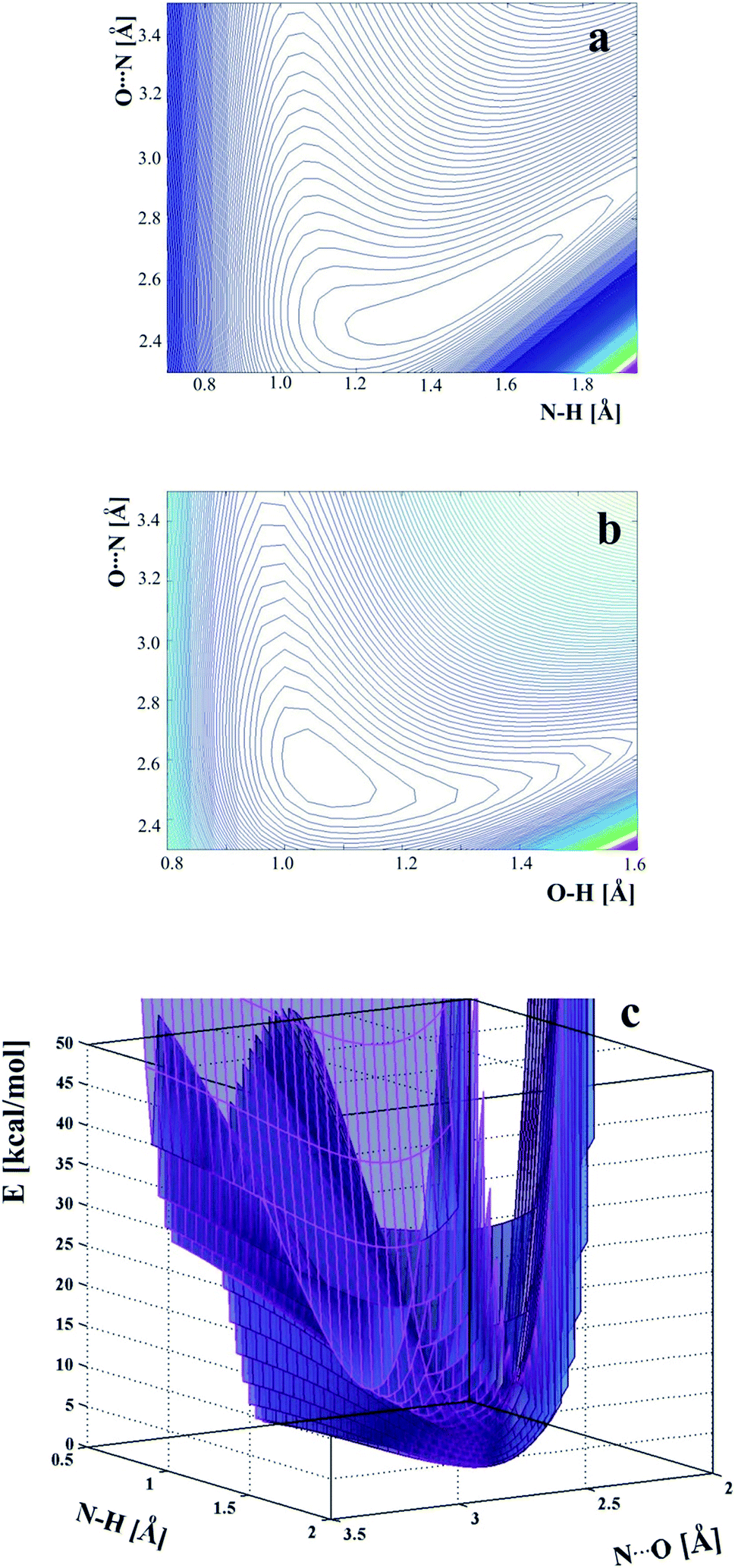 | ||
| Fig. 4 Two-dimensional potential energy surfaces as a function of: (a) – OH and O⋯N distances, (b) – OH and O⋯N, (c) – potential energy surface at the level of the second minimum which spreads in the proton acceptor region. | ||
Comparison of the energy plots in Fig. 4: O⋯N versus OH and O⋯N versus NH shows that it is very easy to change the location of the proton in the hydrogen bridge, but more energy is needed to elongate the O⋯N distance. For this reason, temperature can change the OH and NH bond lengths when the O⋯N distance is the same. The lowest energy is characteristic of the location of the proton at the oxygen atom. The second, very broad minimum appears at the acceptor, and the energy difference between the two minimums is only 2.37 kcal mol−1. The flat shape of the two-dimensional energy surface does not allow a determination of the barrier between the two minimums and illustrates that for a very strong hydrogen bond, each location of the proton along the OHN hydrogen bridge is possible, taking into account the shape of the energy surface.
3. NBO analysis of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate
Molecular electronic density can be described in the frame of Löwdin's concept of Natural Bond Orbital (NBO) which is equivalent to the idealized Lewis structure.33,34 The molecules are composed of atoms linked with bonds expressed as bonding orbitals with localized electrons, nonbonding lone pairs and empty antibonding orbitals.35 If the localized Lewis structure is not perfectly realized, the occupancy of valence antibonding orbitals increases. Linear combination of the parent NBO with participation of the antibonding orbital gives the Natural Localized Molecular Orbital (NLMO). A measure of the realization of the natural Lewis structure is the percentage of the parent NBO in NLMO.NBO analysis performed previously12 for the experimental neutron structures of complex of 3,5-dinitrobenzoic acid with 3,5-dimethylpyridine and pyridinium 2,4-dinitrobenzoate with the proton moving upon temperature change showed significant rearrangement of molecular orbitals caused by the proton shifting in the hydrogen bond. In 3-methylpyridinium 2,6-dichloro-4-nitrophenolate the proton location in the hydrogen bond is sensitive to temperature but moving of the proton is rather limited and, independent of temperature, the hydrogen bond has ionic character. To follow through the proton moving from molecular to proton transfer complexes with particular noticing of the proton close to the hydrogen bond middle, NBO analysis has been performed for theoretical structures with continuous proton moving from the donor to acceptor. Also for the experimental neutron structures the NBO analysis has been done. Investigation of molecular orbitals of the proton donor and proton acceptor explains the mechanism of the proton transfer. The investigated NLMOs are presented in Fig. 5.
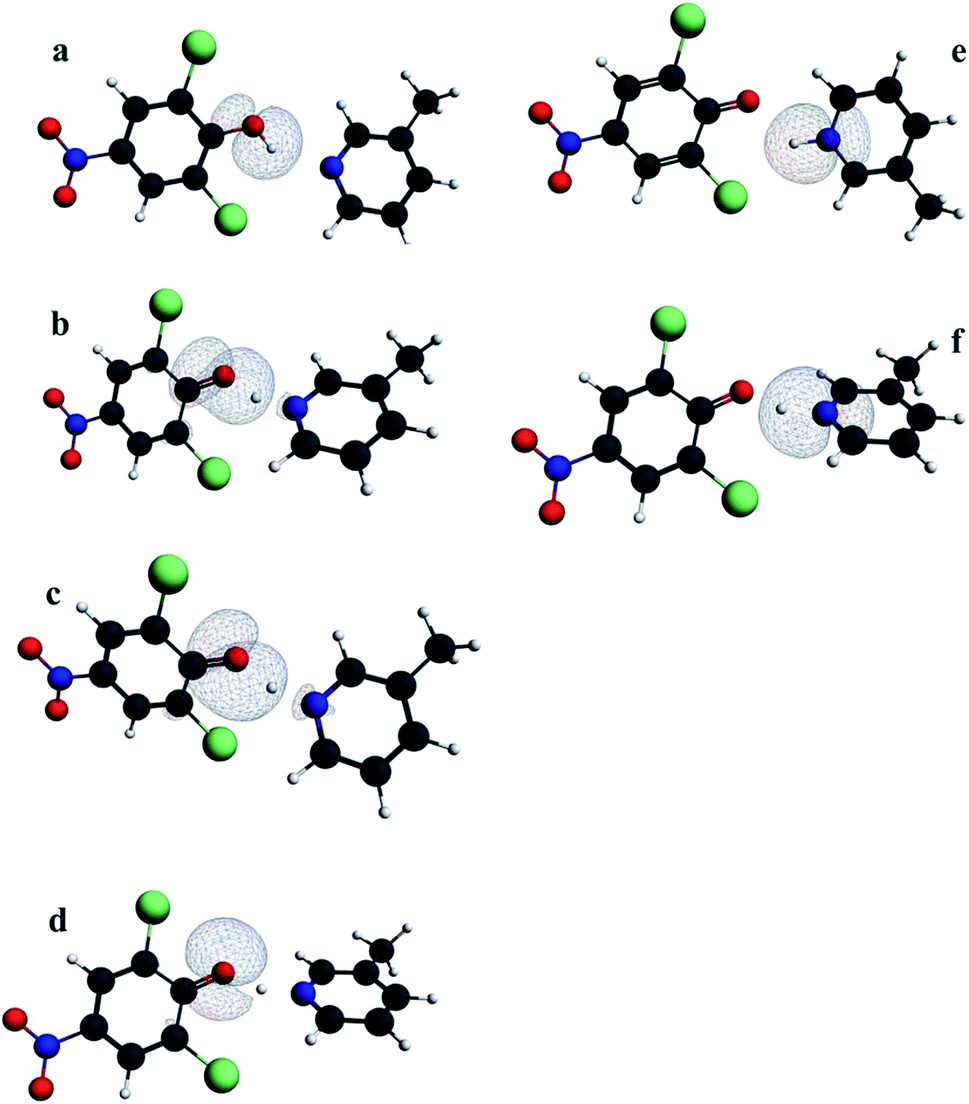 | ||
| Fig. 5 NLMO lone pair orbitals and OH and NH bond orbitals in the proton donor and the proton acceptor in 3-methylpyridinium 2,6-dichloro-4-nitrophenolate. (a) – OH bond, (b–d) – lone pairs of the proton donor, (e) – NH bond, (f) – lone pair at the proton acceptor. The orbital “d” is not sensitive to the proton transfer. | ||
Percentage of the parent NBOs in NLMOs is a measure of delocalization of the orbitals that changes with moving of the proton in the hydrogen bond. Among the orbitals in Fig. 5 only the orbital “d” is insensitive to proton transfer. Other orbitals are strongly delocalized except the OH and NH bonds that exist only for typical molecular or typical ionic hydrogen bonds respectively. For strong hydrogen bonds orbitals representing OH and NH bonds are replaced by delocalized orbital “c” (Fig. 5). It is characteristic that slight elongation of OH bond is sufficient to replace the OH bond orbital with orbital “c” typical for strong hydrogen bond. For the strongest hydrogen bonds with location of the proton in between the donor – acceptor distance, both oxygen lone pairs participating in the proton transfer as well as the lone pair of the acceptor are characterized by similar delocalization. The points representing the experimental structures confirm the theoretical correlations in Fig. 6.
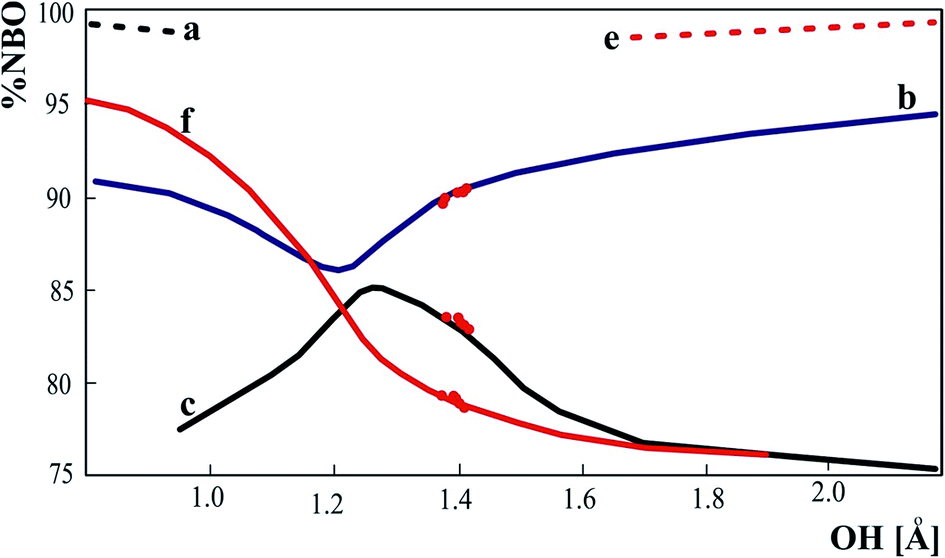 | ||
| Fig. 6 % of the parent NBO orbitals in delocalized NLMO. a, b, c, e, f refers to the orbitals in Fig. 5. The dots represent the experimental neutron structures of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate. | ||
Proton moving in the hydrogen bond is reflected in the participation of the proton in the NLMOs of the donor and acceptor. Percentage of the proton in orbitals presented in Fig. 7 illustrates the mechanism of proton transfer. For the strong hydrogen bond the proton is not engaged in OH or NH bond but participates in donor and acceptor lone pairs. Percentage of proton in NLMO is related to delocalization of the lone pairs and if the molecular orbital is more delocalized, the contribution of the proton is more significant.
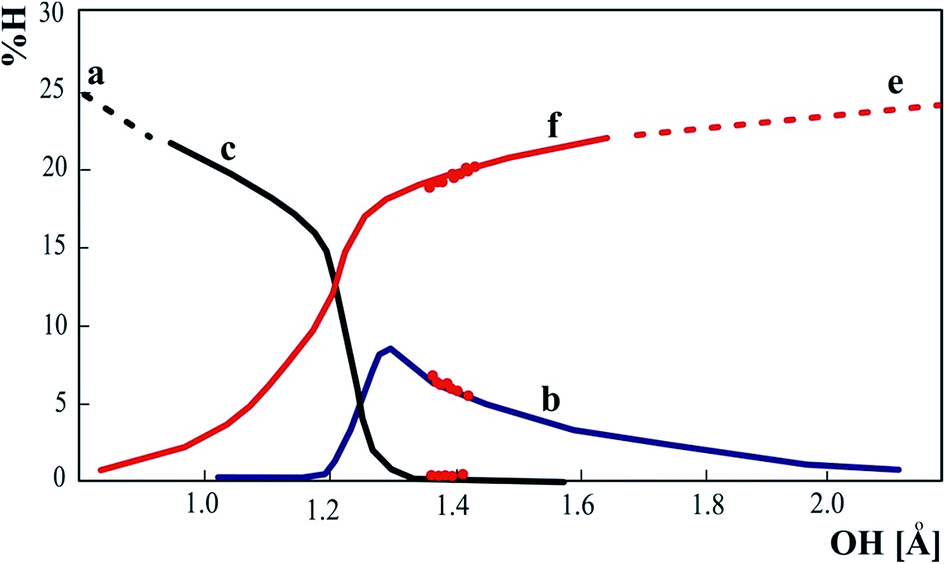 | ||
| Fig. 7 Percentage contribution of proton NBO orbital in delocalized NLMO of donor (a–c) and acceptor (e and f). The dots represent the experimental neutron structures of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate. | ||
Shifting of the proton to the middle of the O⋯N distance is connected with similar delocalization of the molecular orbitals participating in proton transfer so the proton can be similarly engaged in the electron cloud of the proton donor and acceptor.
Limited amount of the experimental structures with the proton moving is not sufficient to follow the proton transfer from donor to acceptor. Combination of the data for experimental structures with correlations obtained theoretically allows investigation of the proton transfer mechanism for complete range of the proton transfer.
4. Comparison of AIM parameters for 3-methylpyridinium 2,6-dichloro-4-nitrophenolate and other OHN intermolecular hydrogen bonded complexes
Theoretical analysis developed by Bader36 provides a picture of the electron cloud, quantitative parameters describing the electron density at the critical points and the parameters of the atomic basins is the method “Atom in Molecules” (AIM) called also “Quantum Theory of Atom in Molecules” (QTAIM). The most important parameters in AIM analysis are connected with the bond critical points (BCP). The existence of BCPs with presence of bond paths linking two atoms is a necessary condition for the existence of a chemical bond. AIM method delivers criteria for existence of the hydrogen bond giving precise values of the electron density at the bond critical point as higher than 0.002 au, that was found to be the boundary value for the existence of a hydrogen bond,37 the Laplacian of electron density more than 0.004 au and the shape of the electron density paths that cannot be bent and characterized by high ellipticity.The eigen values of the Hessian matrix of the electron density at BCP (λ1, λ2, λ3) are a source of information on stability of the bond and energetic properties of the electrons at BCP. The ellipticity (ε = (λ1/λ2) − 1) of electron density at BCP describes the deviation from spherical concentration of the electron density at the BCP. The kinetic and potential energy density can be extrapolated as: G(r)BCP = 15.3λ3 and V(r)BCP = 35.1(λ1 + λ2).38 The potential energy of the electrons at BCP expresses the pressure exerted on the electrons by other electrons; the kinetic energy expresses the mobility of the electrons at the BCP and their pressure exerted on other electrons. The Laplacian of the electron density at BCP can be used to classify the interatomic interactions as the shared-shell (∇2ρ(r) < 0) and the closed-shell interaction (∇2ρ(r) > 0). For the closed shell interaction and the values of |V(r)BCP|/G(r)BCP between 1 and 2 the hydrogen bond has partially covalent character.39
The AIM parameters characterizing the OH and NH bond critical points for the neutron structures of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate are collected in Table 3. In all the analyzed structures, the OHN hydrogen bond is strong so the values of the electron density are significantly higher than the value of 0.002 [a.u.]. Other criteria of hydrogen bond existence are also fulfilled. The values of the electron density, Laplacian, potential, kinetic energy of the electrons at the BCPs of the O–H and N–H bonds indicate existence of strong hydrogen bond.40 The ratio |V(r)BCP|/G(r)BCP for OH bond indicates a noncovalent interaction with participation of covalent character.
| Temp [K] | Bond | ρ(r) | ∇2ρ(r) | G(r)BCP [kJ mol−1] | V(r)BCP [kJ mol−1] | ε |
|---|---|---|---|---|---|---|
| 10 | O–H | 0.0931 | 0.126 | 8.496 | −15.079 | 0.012 |
| N–H | 0.239 | −1.027 | 11.184 | −61.695 | 0.021 | |
| 30 | O–H | 0.094 | 0.121 | 8.580 | −15.437 | 0.014 |
| N–H | 0.235 | −0.981 | 11.142 | −59.979 | 0.021 | |
| 50 | O–H | 0.093 | 0.128 | 8.466 | −14.946 | 0.014 |
| N–H | 0.241 | −1.048 | 11.197 | −62.671 | 0.021 | |
| 80 | O–H | 0.095 | 0.124 | 8.684 | −15.563 | 0.013 |
| N–H | 0.238 | −1.014 | 11.216 | −61.316 | 0.021 | |
| 110 | O–H | 0.091 | 0.134 | 8.395 | −14.563 | 0.012 |
| N–H | 0.248 | −1.119 | 11.310 | −65.230 | 0.020 | |
| 140 | O–H | 0.097 | 0.120 | 8.805 | −15.974 | 0.013 |
| N–H | 0.234 | −0.972 | 11.250 | −59.747 | 0.021 | |
| 170 | O–H | 0.098 | 0.116 | 8.938 | −16.434 | 0.013 |
| N–H | 0.230 | −0.933 | 11.154 | −58.340 | 0.021 | |
| 200 | O–H | 0.098 | 0.119 | 8.908 | −16.272 | 0.012 |
| N–H | 0.232 | −0.950 | 11.146 | −58.915 | 0.021 | |
| 230 | O–H | 0.100 | 0.112 | 9.041 | −16.827 | 0.013 |
| N–H | 0.225 | −0.883 | 11.073 | −56.378 | 0.021 | |
| 260 | O–H | 0.099 | 0.111 | 8.986 | −16.725 | 0.012 |
| N–H | 0.225 | −0.879 | 11.068 | −56.237 | 0.021 | |
| 290 | O–H | 0.103 | 0.098 | 9.244 | −17.750 | 0.012 |
| N–H | 0.216 | −0.792 | 10.978 | −52.976 | 0.021 |
In Fig. 8 is presented dependency of ρ(r)OH and ρ(r)NH on the OH bond length calculated for 3-methylpyridinium 2,6-dichloro-4-nitrophenolate with gradual shift of the proton realized by change of the OH bond length and optimization of other structural parameters. The obtained curves are identical with previously analyzed dependencies of AIM parameters for the complex of trimethylamine with 2,6-dichloro-4-nitrophenol.41 Analogous dependencies for other phenols are sensitive to the proton donor. Replacing of trimethylamine with 3-methylpyridine does not change the correlation of ρ(r)OH and ρ(r)NH on the OH bond length that is in agreement with the higher sensitivity of the proton donor than the proton acceptor to the shifting of the proton.
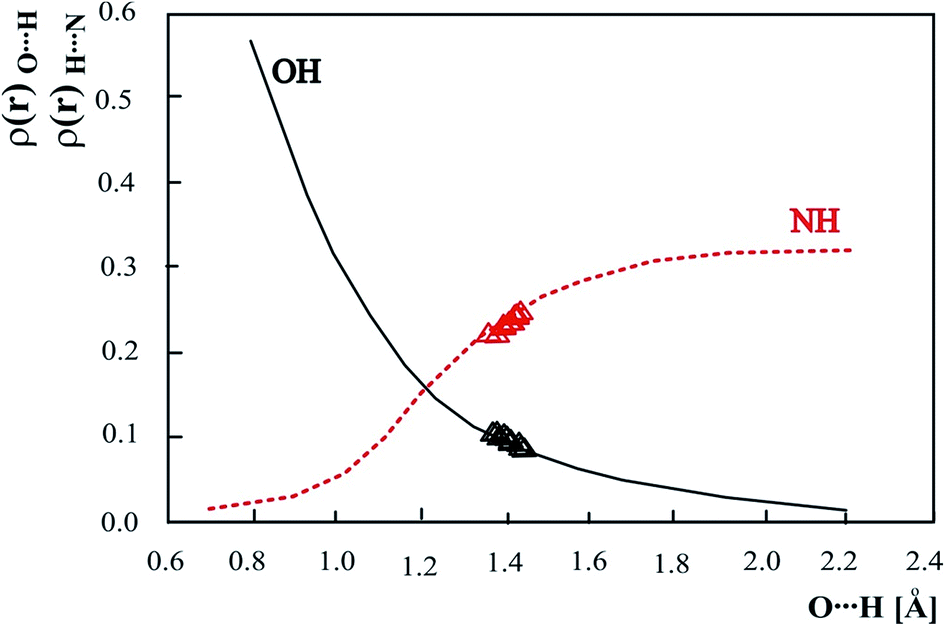 | ||
| Fig. 8 Theoretical electron density (ρ(r)) at the OH and NH (3,−1) critical point as a function of OH distance. The values for experimental neutron structure are marked as Δ. | ||
Electron density values calculated for the experimental neutron structures of 3-methylpyridinium 2,6-dichloro-4-nitrophenolate are located on the curve obtained for the optimized structures. This fact confirms the general character of the dependence of the electron density on the distance common for calculated and experimental results.
In Fig. 9 electron densities at BCP of the OH and NH bonds calculated for all intermolecular OHN neutron structures measured at different temperatures have been compiled. The electron densities at OH and NH BCP have been calculated and correlated with the difference of OH and NH bond length used as a measure of the hydrogen bond strength. For the strongest hydrogen bonds the difference between OH and NH bond length should be close to zero although the crossing point at OH–NH equal −0.0468 Å and the electron density of 0.1579 [a.u.] reflects nonequivalence of OH and NH bonds. The correlations in Fig. 9 include the data for phenols and benzoic acids and are common for OHN complexes independent of the proton donor. Electron densities at bond critical points are common for all OH proton donors and N proton acceptors and insensitive to particular molecules in the intermolecular hydrogen bond.
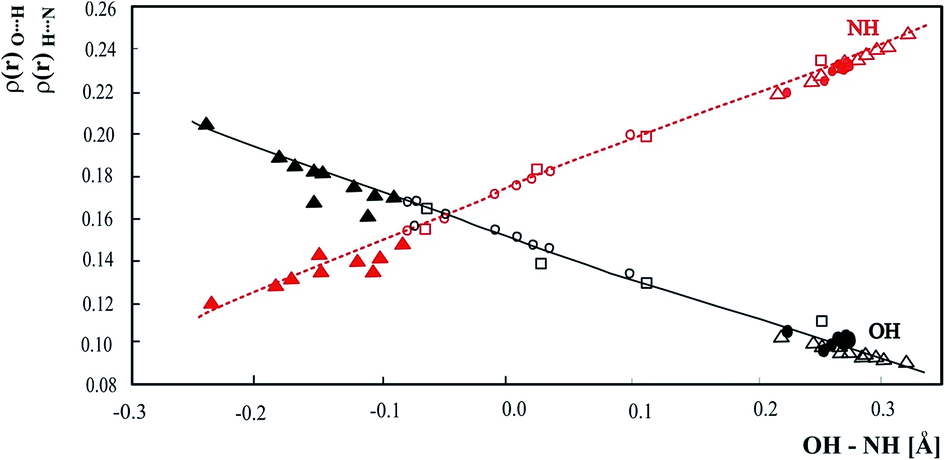 | ||
| Fig. 9 Relationship of ρ(r) at the OH and NH BCPs with OH – NH difference. • – pyridinium 2,4-dinitrobenzoate,27 o – 4-methylpyridine – pentachlorophenol complex,28 □ – 1:2 adduct of benzene-1,2,4,5-tetracarboxylic acid and 4,4′-bipyridyl,29 ▲ – complex of 3,5-dinitrobenzoic acid with 3,5-dimethylpyridine,12 Δ – 3-methylpyridinium 2,6-dichloro-4-nitrophenolate. | ||
Conclusions
The very short hydrogen bond in the 2,6-dichloro-4-nitrophenol – 3-methylpyridine complex belongs to the critical region in which the proton location can be easily modified. The one- and two-dimensional energy surfaces calculated using solid state geometry are very flat, and even low energy is sufficient to move the proton along the hydrogen bridge. More energy is needed to elongate the O⋯N hydrogen bridge. The investigated structures with precise proton location in the hydrogen bond show that in strong hydrogen bond proton is not necessary located close to the middle of the O⋯N distance, but because of very low energy barrier between two minimums on the potential energy surface every proton location is possible. In strong hydrogen bond central location of the proton is connected with delocalization of the orbitals of donor and acceptor and similar participation of proton in donor and acceptor NLMOs.Acknowledgements
The Wroclaw Center for Networking and Supercomputing is acknowledged for generous computer time. Experiments at the ISIS Neutron and Muon Source were supported by a beamtime allocation from the Science and Technology Facilities Council.References
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1990 Search PubMed.
- D. Hadži, Hydrogen Bonding, Pergamon Press, 1969 Search PubMed; D. Hadži, Theoretical Treatment of Hydrogen Bonding, J. Wiley & Sons, 1997 Search PubMed.
- J. J. Valentini, M. J. Coggiola and Y. T. Lee, J. Am. Chem. Soc., 1976, 98, 854 CrossRef.
- P. Huyskens and T. Zeegers-Huyskens, J. Chim. Phys., 1964, 61, 81 Search PubMed; H. Ratajczak and L. Sobczyk, J. Chem. Phys., 1969, 50, 556 CrossRef CAS.
- R. Lindemann and G. Zundel, J. Chem. Soc., Faraday Trans. 2, 1977, 73, 788 RSC; G. Albert and G. Zundel, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 553 RSC; R. Janoschek, E. G. Weidemann, H. Pfeiffer and G. Zundel, J. Am. Chem. Soc., 1972, 94, 2387 CrossRef CAS; U. Böhner and G. Zundel, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 1425 RSC.
- I. Olovsson and P.-G. Jönsson, in The Hydrogen Bond – Recent Developments in Theory and Experiments, ed. P. Schuster, G. Zundel and C. Sandorfy, Amsterdam, North-Holland, 1976, pp. 426–433 CrossRef CAS; T. Steiner and W. Saenger, Acta Crystallogr., Sect. B: Struct. Sci, 1994, 50, 348 CrossRef CAS; T. Steiner, J. Chem. Soc., Chem. Commun., 1995, 1331 RSC; T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef.
- J. Hawrnek, J. Oszust and L. Sobczyk, J. Phys. Chem., 1972, 76, 2112 CrossRef CAS; J. Jadzyn and J. Małecki, Acta Phys. Pol., 1972, 41, 599 Search PubMed; P. Huyskens, W. Cleuren, M. Franz and M. A. Vuylsteke, J. Phys. Chem., 1980, 84, 2748 CrossRef.
- A. Rabold and G. Zundel, J. Phys. Chem., 1995, 99, 12158 CrossRef CAS; Z. Malarski, M. Rospenk, L. Sobczyk and E. Grech, J. Phys. Chem., 1982, 86, 401 CrossRef; C. L. Bell and C. M. Barrow, J. Chem. Phys., 1952, 31, 1158 CrossRef; E. Grech, J. Kalenik, Z. Malarski and L. Sobczyk, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2005 RSC.
- E. Kwiatkowska, I. Majerz and A. Koll, Chem. Phys. Lett., 2004, 398, 130 CrossRef CAS.
- I. Majerz and A. Koll, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 406–415 CrossRef CAS PubMed.
- C. C. Wilson, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 435 Search PubMed; C. C. Wilson, K. Shankland and N. Z. Shankland, Kristallografiya, 2001, 216, 303 Search PubMed; E. C. Kostanek and W. R. Busing, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 2454 CrossRef CAS; B. L. Rodrigues, R. Tellgreen and N. G. Fernandes, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 353 Search PubMed; I. Olovsson, H. Ptasiewicz-Bąk, T. Gustafsson and I. Majerz, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 311 Search PubMed.
- I. Majerz and M. J. Gutmann, RSC Adv., 2011, 1, 21 RSC.
- I. Majerz, W. Sawka-Dobrowolska and L. Sobczyk, J. Mol. Struct., 1993, 279, 177 CrossRef.
- D. A. Keen, M. J. Gutmann and C. C. Wilson, J. Appl. Crystallogr., 2006, 39, 714 CrossRef CAS.
- M. J. Gutmann, SXD2001, ISIS Facility, Rutherford Appleton Laboratory, Oxfordshire England, 2005 Search PubMed.
- C. C. Wilson, J. Appl. Crystallogr., 1997, 30, 184 CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 09, Revision A.9, Gaussian, Inc., Pittsburgh PA, 2009 Search PubMed.
- J. Stare, J. Mavri, G. Ambrožič and D. Hadži, J. Mol. Struct., 2000, 500, 429 CrossRef CAS.
- J. Stare and J. Mavri, Comput. Phys. Commun., 2002, 143, 222 CrossRef CAS.
- NBO 5.0. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001, http://www.chem.wisc.edu/%7Enbo5.
- F. Biegler-König, J. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545 CrossRef.
- T. Steiner, J. Phys. Chem. A, 1998, 102, 7041 CrossRef CAS.
- H.-H. Limbach, M. Pietrzak, S. Sharif, P. M. Tolstoy, I. G. Shenderovich, S. N. Smirnov, N. S. Golubev and G. S. Denisov, Chem.–Eur. J., 2004, 10, 5195 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542 CrossRef CAS.
- X.-Z. Li, B. Walker and A. Michaelides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6369 CrossRef CAS.
- I. Majerz and M. J. Gutmann, J. Phys. Chem. A, 2008, 112, 9801 CrossRef CAS PubMed.
- T. Steiner, I. Majerz and C. C. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2651 CrossRef CAS.
- J. A. Cowan, J. A. K. Howard, G. J. McIntre, S. M.-F. Lo and J. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 794 Search PubMed.
- J. A. Cowan, J. A. K. Howard, G. J. McIntre, S. M.-F. Lo and J. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 724 Search PubMed.
- I. Majerz, Z. Malarski and L. Sobczyk, Chem. Phys. Lett., 1992, 274, 361 CrossRef.
- J. Li, X. Li, S. S. Iyengar and J. Chem, J. Chem. Theory Comput., 2014, 10, 153 CrossRef.
- A. E. Reed, L. A. Cyrtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762 CrossRef CAS.
- F. Weinhold and C. R. Landis, Chem. Educ.: Res. Pract. Eur., 2001, 2, 91 RSC.
- R. F. W. Bader, Atoms in Molecules: A quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem. A, 1995, 99, 9747 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS; E. Espinosa, C. Lecomte and E. Molins, Chem. Phys. Lett., 1999, 300, 745 CrossRef.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS; Y. A. Abramov, Acta Crystallogr., Sect. A: Found. Crystallogr., 1997, 53, 264 CrossRef.
- S. J. Grabowski, W. A. Sokalski and J. Leszczyński, J. Phys. Chem. A, 2006, 110, 4772 CrossRef CAS PubMed.
- E. Kwiatkowska and I. Majerz, J. Phys. Org. Chem., 2008, 21, 867 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1060082–1060092. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra06733b |
| This journal is © The Royal Society of Chemistry 2015 |