
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Validating potential energy surfaces for classical trajectory calculations
Huw O. Pritchard*
Department of Chemistry, York University, Toronto, Canada M3J 1P3. E-mail: huw@yorku.ca
First published on 10th July 2015
Abstract
Two possible methods for probing flaws in empirical potential energy surfaces are examined, using a simple unimolecular isomerisation as an example. One is a heuristic estimate of the degree of chaos in a vibrating molecule; the other is the energy distribution profiles at the instant of reaction. It is found that deliberate increases in the degree of computational chaos do not alter the ensemble average of intrinsic vibrational chaos in these trajectories.
Introduction
Since the advent of Eyring–Polanyi transition state theory, it has seemed almost axiomatic that a classical trajectory calculation for a unimolecular reaction on a carefully constructed potential energy (PE) surface could yield an acceptable reaction rate; this assumption rests primarily on the notion that classical and quantum properties converge at high energy, i.e. the correspondence principle will hold for the vibrational motions of a polyatomic molecule, which seems to be a reasonable assumption.1 The discussion here will be confined to unimolecular reactions, but these considerations apply equally to bimolecular reactions, and beyond.2Hundreds of thermal unimolecular reactions have been reported over the past 60 years;3 among those, three of the most extensively studied, the isomerisations of methyl isocyanide,4,5 of cyclopropane,6 and the thermal dissociation of di-tert-butyl peroxide,7 present clear challenges for computational modelling.
At the present time, ab initio potential energy surfaces are not available for any of these reactions, but for the simplest case, CH3NC ⇌ CH3CN, one can be expected before long. For the other two target reactions, an ab initio potential energy surface for the isomerisation of cyclopropane to propylene may follow later, but probably not for the thermal dissociation of di-tert-butyl peroxide, for which one would need to create an empirical surface. But, as Bowman et al.8 have said, empirical surfaces are “highly problematic”, and the purpose of the work described below is to probe possible methods for exploring deficiencies.
In fact, such deficiencies arise in the simplest of these three cases, that of classical trajectory calculations for the reaction CH3NC ⇌ CH3CN using the well-known empirical potential energy surface of Sumpter and Thompson.9 There, the asymptotic forms at large and small interatomic distances are realistic, and the vibrational properties near the equilibrium structures are well represented. Also, transition state theory (TST) calculations using these parameters yield infinite-pressure rate constants close to the observed values between 400 and 600 K.10 In stark contrast, trajectory methods using this same PE surface yield reaction rates about a factor of 20 below the experimental values, with the forward and reverse rates failing to obey microscopic reversibility.11–13 As TST is an equilibrium theory,14 it cannot illuminate dynamical properties of the reacting molecules in ways that trajectory methods do, and so these conflicts need to be understood.
An earlier study12 had confirmed that a similar calculation for the reaction NCNC ⇌ NCCN, but using an ab initio potential energy surface, gave results consistent with microscopic reversibility, implying that the CH3NC ⇌ CH3CN potential energy surface must be defective somewhere in an intermediate energy region. It had long been suspected that the cause may be a lack of sufficient vibrational mixing in this energy range,11 and led to my development and initial testing of a speculative method for quantifying the degree of chaos in a vibrating molecule.13 This involved the derivation of a “mechanical” spectrum as the Fourier transform of the sum of all inter-atomic distances in the vibrating molecule; this type of spectrum should not be confused with a conventional experimental electromagnetic spectrum. Some features of such a spectrum could be identified by comparison with transforms of individual distances, or of groups of distances, and proved to be helpful in the analysis of the present calculations.
Most of the emphasis of this paper is focussed on the CH3NC molecule, but this potential energy surface has its origin at the potential minimum of CH3CN. As noted previously,13 the electron-volt (1 eV = 23.06 kcal mol−1 = 96.5 kJ mol−1 = 8066 cm−1) is a useful energy unit as it puts the potential minimum of CH3NC at 1.0 eV with respect to the global minimum. The zero-point energy (zpe = 9626 cm−1 = 1.19 eV) of CH3NC then stands at 2.1980 ⋍ 2.2 eV, and the potential barrier at ∼2.8 eV; the zero-point energy of CH3CN (9623 cm−1) is also 1.19 eV, but is not used in the present work.
Anchoring the energy measurements to the minimum of the CH3CN system can lead to confusion, but I have tried to make this distinction clear all through. Energies in eV are measured from the potential minimum of CH3CN unless otherwise stated, with an explanatory comment if needed; wavenumbers will be used mainly for state densities and spectral properties, and values imported from elsewhere are cited in their original units.
Table 1 lists a selection of relevant data for the forward reaction CH3NC → CH3CN from earlier calculations,11 to provide some justification for the classical approach. It shows that a quantum state having any of these lifetimes would overlap with thousands of neighbouring states,15 so that its trajectory may explore the whole of the available phase space within that energy band. These energies start somewhat above reaction threshold (the experimental activation energy16 is 38.2 kcal mol−1, i.e. 1.66 eV above zpe, 3.86 eV above zero), but as these calculated lifetimes are more than an order of magnitude too large it is safe to accept that the natural width of a transitioning state near threshold will still far exceed the spacing between the states. Hence, the motions in the thermal reaction range can be assumed to be chaotic, and a classical simulation should mimic the true behaviour.
| Energy above zero [eV] | Energy above zpe [cm−1] | ρ(E)NC states per cm−1 | τNC [ps] | Natural width [cm−1] |
|---|---|---|---|---|
| 4.3 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 019 019 |
9.6 × 104 | 203 | ∼0.15 |
| 5.3 | 33085 |
9.8 × 105 | 55 | ∼0.6 |
| 6.3 | 41150 |
6.7 × 106 | 16 | ∼2 |
Were this not to be so, Baer and Hase17 consider a limiting case where the phase space is divided into two regions, “one part consisting of chaotic trajectories which can decompose and the other of quasi-periodic trajectories which are trapped in the reactant phase”. They then introduce the ratio fch = ρch(E)/ρ(E), being the fraction of phase space that supports chaotic motion, and defines the amount by which the computed classical rate would fall below the true value.
Computational procedure
For a CH3NC or CH3CN molecule in its normal internuclear configuration, each of the 6 atoms is assigned a set of random momenta in the x, y, and z directions; any unwanted translation and rotation are removed, and the motions scaled to the target energy value. Then, for each initial energy distribution, after a simple fourth-order Runge–Kutta start-up procedure, the trajectory is followed by a fifth-order Adams–Mouton predictor-corrector. For each trajectory, a standard time step length of 0.1 fs was used, and arithmetic sums of all N values of internuclear displacements i.e.were tabulated at δt = 2.5 fs intervals;† for some applications, a vector P(t) of the instantaneous potential energy values was also saved. Throughout this study, the minimum ensemble size was 1000 trajectories. These methods have been described extensively in previous publications,12,13 and all algorithms used are described in detail elsewhere.18
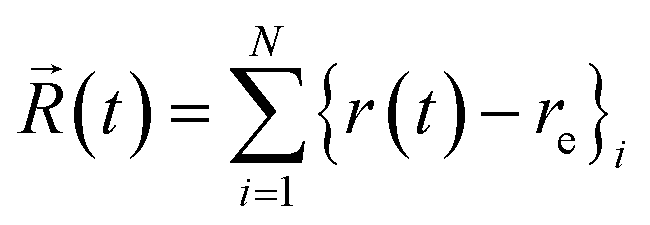
In the preceding paper,13 I speculated that a possible surrogate for fch can be obtained from the spectral properties of such classical trajectories. The Fourier transform of the vector ![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0052_20d1.gif) (t) yields an overall spectrum of the collective vibrations. The spectral resolution is determined by the vector length, and with 32768 (215) terms yields a resolution of about 6.5 cm−1, thus requiring trajectory lengths of 81.92 ps. The area under each spectral curve is a measure of the total spectral intensity and rises linearly with increasing energy above the classical minimum.13 The postulate then is that the fraction of the total intensity lying below the lowest vibrational frequency, 263 cm−1 for CH3NC,19 gives a “measure” of the degree of chaos in the system (and for simplicity is taken to be the area between 0 and 250 cm−1, expressed as a percentage, and called ϕc). That is, if the area under a regular spectrum is proportional to the total vibrational energy, then any energy that is not “seen” by the Fourier transform must be chaotic. As the degree of chaos increases, and the motions degenerate into white noise, for which the Fourier transform is flat, the spectral features shrink and the missing energy migrates to a singularity at zero frequency; the properties of this “spike” are that the greater the number of points in the given frequency range, the narrower and taller this feature becomes, tending to infinity in the limit.20
(t) yields an overall spectrum of the collective vibrations. The spectral resolution is determined by the vector length, and with 32768 (215) terms yields a resolution of about 6.5 cm−1, thus requiring trajectory lengths of 81.92 ps. The area under each spectral curve is a measure of the total spectral intensity and rises linearly with increasing energy above the classical minimum.13 The postulate then is that the fraction of the total intensity lying below the lowest vibrational frequency, 263 cm−1 for CH3NC,19 gives a “measure” of the degree of chaos in the system (and for simplicity is taken to be the area between 0 and 250 cm−1, expressed as a percentage, and called ϕc). That is, if the area under a regular spectrum is proportional to the total vibrational energy, then any energy that is not “seen” by the Fourier transform must be chaotic. As the degree of chaos increases, and the motions degenerate into white noise, for which the Fourier transform is flat, the spectral features shrink and the missing energy migrates to a singularity at zero frequency; the properties of this “spike” are that the greater the number of points in the given frequency range, the narrower and taller this feature becomes, tending to infinity in the limit.20
This behaviour is illustrated in Fig. 1: part (a) shows a typical spectrum for a trajectory that does not isomerise and by comparison with Fig. 3 of the earlier paper,13 which shows the transforms of the 3 C–H or the 3 H–H distances, these features are related to the motions within the CH3 group and are still partially regular; part (b) shows another trajectory, at the same energy, where the spectrum is flat. Instead of terminating this trajectory at the first crossing, it was allowed to run on to 81.92 ps, whence it made 37 crossings, sharing its time between CH3NC and CH3CN; the crossings occur at random potential energy values between 2.9 and 5.4 eV, so that each newly formed isomer evolves from a totally different set of positions and momenta, giving quasi-periodic snippets strung together in random fashion, ensuring the illusion of complete chaos. Also, as dots, is the same spectrum expanded by a factor of 5000, where the remnants of the CH3NC and CH3CN spectra still persist. This is a much stronger demonstration of a chaotic spectrum than found previously.13
 | ||
| Fig. 1 Total spectra for two CH3NC trajectories at 5.1 eV above zpe (total energy 7.3 eV). The percentage intensities (ϕc) below 250 cm−1 are: (a) 15.0%, (b) >99.9%; in panel (b), the signal is also shown at 5000 times magnification. | ||
Fig. 2 shows the behaviour of these ϕc values for CH3NC from an energy of 2 eV above the global minimum (in the classical region, just below the zpe) up to 7.3 eV (zpe + 5.1 eV), at which point, only 20 out of 1500 trajectories survive for 81.92 ps; this is an exceedingly high energy, being 115 kcal mol−1 above the zpe whereas the activation energy is only 38.2 kcal mol−1.16 As these trajectories are only partially chaotic, there is no reason for these chaos fractions, ϕc, at any energy to coincide since each trajectory starts from a unique energy distribution and retains some memory of it.
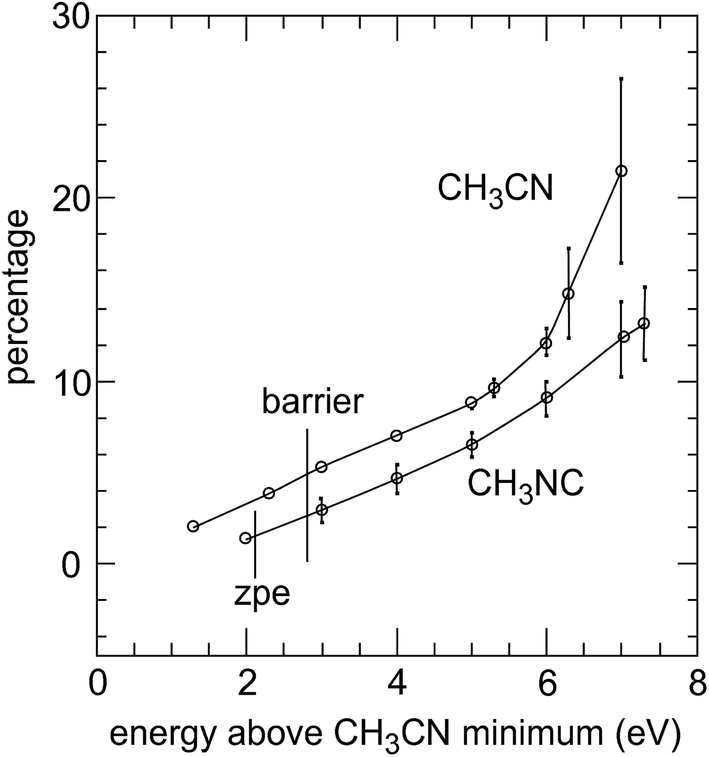 | ||
| Fig. 2 Percentage of spectral intensity residing below 250 cm−1 versus energy above potential minimum; the vertical bars on each point represent the standard deviations from a random selection of 20 trajectories. The position of the barrier for both reactions, and for the zpe of CH3NC are shown by vertical lines; the graph for CH3CN starts at the zpe. | ||
Fig. 2 also shows the corresponding curve for CH3CN, starting at its zero-point energy; up until a total energy of 5 eV, the pattern is very similar to that for CH3NC, but with a downward shift of about 1 eV, the difference in energy of the two systems. Beyond that, there a sharp turn upwards, but it was not possible to collect a set of comparable values at the highest energy as out of 5000 trajectories only one lived for 81.92 ps.
The standard method
The trajectory observations are very similar to those reported previously,21 but then trajectory lengths were restricted (by a CPU speed of 1 MHz) to 3 ps, whereas now, 1 ns is readily accessible. Much of the analysis in this Section uses trajectories for energies of 4.0 and 5.0 eV above the CH3NC potential minimum because, as just noted, too few trajectories survive for the required time at higher energies; conversely, as the energy is reduced, fewer trajectories react, and eventually, none at all.As we,21 and others,22 found, there is a sharp drop within the first ps in the number of molecules remaining, due to some possessing excess energy in the critical directions reacting before equilibration could occur. Then (at a similar energy), a long period, up to over 200 ps (instead of the original 3 ps)21 when the logarithm of the number remaining is closely linear with respect to time; after that, the points become rather scattered, as those few of the original 1000 now left no longer comprise an acceptable statistical sample.
Although it may seem plausible, the lifetime of a trajectory at any given energy does not correlate with its degree of chaos, ϕc: for example, at 4.0 eV above the CH3NC potential minimum, the average ϕc for trajectories with lifetimes between 89 to 222 ps is 6.6% and for 275 to 485 ps, it is 6.5%.
It should also be noted that the spectrum shown in Fig. 1(a) and all those used in the calculation of ϕc are different from the composite spectra shown by Sewell and Thompson:23 those are the sum of the Fourier transforms of each bond distance, whereas these are the Fourier transform of the sums of all inter-atom distances; these are not the same although, often, they can look similar if long-range interactions are unimportant.
Thus, we have a first-order decay process, giving the appearance of a conventional unimolecular reaction,17,24 although none of these single-pass trajectories (as in Fig. 1(a) and similar ones at all lower energies) is anywhere close to being chaotic; otherwise, these spectra would be flat. As seen in Fig. 2, the prospective measure, ϕc, of the degree of chaos rises from near zero at low energy to between 5% near the activation region and 10–15% at unrealistically high degrees of excitation.
First, we might consider this as an extreme limiting case of “intrinsic non-RRKM behaviour”, as depicted in Fig. 1(e) of Bunker and Hase,24 and discussed in more detail by Baer and Hase later.17 After the initial loss of specifically excited molecules during the first ps, we are left with two non-interacting regions of phase space, one whose properties are unknown and does not decay, and one which is chaotic and decays in the expected manner; this could account for the very low rate constant.
In fact, we find that the whole CH3NC ensemble reacts before 385 ps at 5.0 eV, or before 485 ps at 4.0 eV above its minimum. Now add to Fig. 1(e) a slow leakage between the chaotic and the quasi-periodic regions, which Bunker and Hase actually show in their Fig. 4(c),24 but never discuss. From the manner in which the initial energy is allocated to the molecules, very crudely, about two-thirds of the total momentum will reside in the CH3 moiety. During the first ps,21,22 every molecule assigned sufficient N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C angular motion will react, leaving a sub-ensemble of molecules with mainly C–NC vibration and a modicum of C–NC bending motions which are isolated from the motions inside the CH3 group. The excess energy in the methyl group then leaks rather slowly (on a roughly ∼100 ps time scale) by a first order process whence the NC group attains sufficient angular motion to invert almost instantly. Because of the large difference between the rates of these two processes, the population versus time plot in Fig. 1(e) collapses into bimodal form, a sharp initial drop followed by a first order decay. While originally these calculations were assumed to yield a reaction rate,21 all that was being observed was a spurious internal relaxation!
C angular motion will react, leaving a sub-ensemble of molecules with mainly C–NC vibration and a modicum of C–NC bending motions which are isolated from the motions inside the CH3 group. The excess energy in the methyl group then leaks rather slowly (on a roughly ∼100 ps time scale) by a first order process whence the NC group attains sufficient angular motion to invert almost instantly. Because of the large difference between the rates of these two processes, the population versus time plot in Fig. 1(e) collapses into bimodal form, a sharp initial drop followed by a first order decay. While originally these calculations were assumed to yield a reaction rate,21 all that was being observed was a spurious internal relaxation!
The disparity between the calculated and the measured rates is shown in Fig. 3, the solid line being an estimate of the specific rate function, k(E), derived as the Slater–Forst inversion of the experimental Arrhenius plot,25 and the bottom set of discrete points being the calculated results; the open circles are the new and the small solid squares the earlier10 results with, respectively, 1000- and 100-trajectory ensembles.
 | ||
| Fig. 3 Computed specific rate function for CH3NC → CH3CN versus energy above zpe. Line – Slater–Forst inversion of experimental Arrhenius rate law; circles – plain calculation; open squares – 4-atom reversals at 0.1 ps intervals; crosses – open circles scaled by ϕc. Solid squares and triangles are, respectively, plain and 4-atom reversal results from earlier calculations.10 | ||
Further analysis
It is not clear what causes this difficulty but the question should be resolved for this particular reaction once an ab initio potential energy surface becomes available. But, for now, it is easy to rule out the computational method, or any lack of numerical precision, as the cause of this huge degree of disagreement.Throughout the history of such calculations, it has been known that the precision of the computed trajectories has seemed almost irrelevant. Hase and Buckowski26 used the reversal of trajectories to test whether they would return to the original starting configuration, which they did not, and we have demonstrated many examples of vastly different individual trajectory lifetimes with the same algorithm on different machines, or different operating systems on the same machine;12,27 despite such variation in lifetime for the same trajectory, sometimes in excess of a factor of 200, the ensemble lifetime averages agreed well, even for quite small ensembles.26,27 What is important in these calculations is not precision in following the trajectory itself, only that the trajectory should be able to go anywhere within the allowable phase space as long as the energy and angular momentum are conserved; this is where precision is vital.12
So the flaw must lie with the potential energy surface, despite its apparent well-behaved properties. For example, it may be that although this surface conforms to all the parameters, both at the normal structure and near the asymptotic limits, there may be some intermediate impediment that tends to impose “intrinsic non-RRKM behaviour” on the computed solution. It may be that the surface enforces too much quasi-periodic motion within the CH3 group, and thus an insufficient degree of chaos; this would be consistent with ϕc values of the order 3–5% in Fig. 2 and the deficit of a factor of around 20 in the calculated k(E) values over the actual experimental energy range, in Fig. 3.
One proposal for increasing the degree of chaos in a trajectory was to make deliberate discontinuities in the integration paths, chosen so as not to compromise the conservation of energy. A simple way is to reverse the momenta of a few chosen particles at regular time intervals, e.g. 0.1 ps,10 but this only conserves the energy, and not the angular momentum unless it is zero; however, methods for conserving both have been explored previously.28 In this way, the computed k(E) value for the CH3NC → CH3CN reaction was raised by a factor of about 15 near reaction threshold, and of about 5 at very high energy.10 In that study, only reversals of peripheral atoms were used, the N atom and either one, or all three, of the H atoms, and the latter was by far the more effective in accelerating the reaction rate. Here, not every permutation of possible groupings was examined, but it appears that the original 4-atom switch (i.e. N and all H atoms) is the most effective. As an aside, reversal of all 6 atoms results in no reaction, as the system meanders about the initial reversal point, and only drifts away slowly due to rounding errors in the 16th decimal place; also, reversals of 5 atoms each time, or of two groups of 3 atoms alternately, are less effective.
The results of this reversal process are also shown in Fig. 3 as open squares: at high energy, they agree well with the older ones,10 shown as solid triangles, but with a steadily increasing difference at lower energy.‡
In contrast, the mean lifetimes for the reverse reaction CH3CN → CH3NC at the two highest energies are unaffected by the reversal procedure; at lower energies there is a small acceleration, suggesting that impediments to reaction occur mainly on the CH3NC side of the potential energy surface. This observation also helps to resolve the microscopic reversibility problem: the approximate state densities at 41150 cm−1 above the global minimum are12 ρCN = 2.4 × 107 and ρNC = 6.7 × 106 states per cm−1 respectively, then ρCN/ρNC ≃ 3.6 which should be equal to τCN/τ*NC = 8.1 (ps)/3.3 (ps) ≃ 2.5; here, the asterisk denotes the reversal procedure and given the uncertainties of at least ±5% in the 1000-ensemble lifetimes and of those in the state densities, this agreement is promising.
Finally, if the original data (open circles in Fig. 3) are scaled by the corresponding value of ϕc as suggested by Baer and Hase,17 we get τ×NC = τNC/ϕc, depicted as crosses in Fig. 3. The τ×NC values track the τ*NC points fairly closely, giving τCN/τ×NC = 8.1 (ps)/2.4 (ps) ≃ 3.4, but detailed analysis is not yet warranted because of the crudity of the state-density estimates; at this point, an exact count would be needed, together with a detailed analysis of the anharmonicities of all the vibrations in both molecules.
Summary of reversal patterns
In this Section, we need to calculate several thousand trajectories to illustrate a single item: hence, most of the following discussion is confined to higher energies, principally 7.0 eV above the CH3CN minimum (6.0 eV above the CH3NC minimum).(i) Without reversals: Fig. 4 shows the average distribution of potential energy for both CH3NC and CH3CN at a total energy E = 7.0 eV. The upper histograms give plots of the vector P(t), with the instantaneous potential energy values sorted into 0.1 eV bins. As we have shown previously,11 the potential energy distribution for a collection of non-interacting harmonic oscillators can be represented by
| f(P) = const × sinm(πP) |
 | ||
| Fig. 4 Potential energy distributions for a total energy of 7.0 eV. The large top plots are histograms of the 32768 energy values P(t); the small lower plots are histograms of the potential energy crossing values for 2000 trajectories each, and the lowest one in (b) is copied from panel (a) to demonstrate their equivalence. The smooth curves are fitted sinm(πP) functions with m = 4.3 or m = 12, as described in the text. | ||
Also, in both of these diagrams, downward-pointing zpe markers are placed at 1.2 eV above their respective minima, showing that either molecule spends very little time in the forbidden region below the zero-point energy; however, this fraction increases rapidly as the total energy is reduced, but once the value of m for the potential energy surface being studied is known, the fraction is easily estimated with the above formula.
The lower histogram in Fig. 4(a) is that of the potential energies at which 2000 trajectories crossed from CH3NC to CH3CN; the vertical markers indicate the range over which P is scaled, the height of the transition barrier (2.8 eV), and the total energy E (7.0 eV). In Fig. 4(b), the corresponding histogram is that for the opposite reaction, and the bottom curve is a copy (slightly displaced) of that from Fig. 4(a) showing that these two profiles are the same. This observation implies, importantly, that if neither set of molecules has any impediment in accessing the transition region, microscopic reversibility will be obeyed.
The most puzzling aspect of these two reaction potential energy profiles is that they, too, conform to a sinm(πP) shape, but the best fit is for m ≃ 12, which happens to be the number of oscillators in each system. However, for n uncoupled oscillators,11
| m ≃ (0.8n − 0.7), n ≥ 4 |
(ii) With reversals: Fig. 5(a) and (b) show corresponding graphs for trajectory ensembles in which the motions of the N atom and all three H atoms were reversed every 0.1 ps. Now, the potential energy distribution functions exhibit the same values of m (4.3 and 12) as do the unperturbed histograms in Fig. 4, but all the curves are shrunk downwards in energy by ∼0.5 eV compared with the sinm(πP) functions copied from Fig. 4. It appears that, somehow, this procedure prevents access to the topmost energy range.
 | ||
| Fig. 5 Effect of equally-spaced reversals on the potential energy distributions, P(t), for a total energy of 7.0 eV. The smooth curves are copied from Fig. 4(a) and (b) respectively, and the two pairs of histograms show a shift downwards of ∼0.5 eV in each case. The lowest histogram in panel (a) shows the crossing energies of 2000 trajectories with randomly-spaced reversals; as in Fig. 4, all fitted sinm(πP) curves have either m = 4.3 or m = 12. | ||
More detailed examination reveals, in fact, that the reversal procedure does not alter the degree of chaos in these systems. If the degree of vibrational mixing, and therefore chaos, were to be increased by the reversal process, then the value of m fitting the overall potential energy distribution would be smaller in Fig. 5 than in Fig. 4.11 Moreover, the values of ϕc found in the Fourier transform spectra of (t) remain within the same bounds as in Fig. 2, with or without reversals. Clearly, the introduction of additional computational chaos does not increase the degree of vibrational chaos; it seems that it must simply increase the chance of the trajectory finding itself in the vicinity of the transition region. However, how it reduces the mean potential energy of the system, and of the reacting molecules, is not obvious.
Fig. 5(a) includes an extra variation, that of random reversal spacings in the range 0.1–0.2 ps, to eliminate the possibility of a spectral feature related to the reversal frequency, but none was found. Both the overall potential energy and the reaction energy profile (bottom line) remained the same, but the mean lifetime was about 15% longer, possibly because the mean time between reversals was now 0.15 ps instead of 0.1 ps, with a concomitant reduction in computational chaos.
A number of mainly negative conclusions can be deduced from these two pairs of diagrams. For both CH3NC and CH3CN, the reversal procedure causes the overall PE and the reactive PE curves to squeeze down to lower energy by about 0.5 eV; in case Fig. 5(a), the mean lifetime at total energy 7.0 eV is reduced from 20.8 to 3.7 ps, but in case Fig. 5(b) only from 11.6 to 11.5 ps. As the relative shapes and positions of the normal and reactive PE profiles hardly change, the large lifetime differences do not depend directly on these factors.
Conclusions
Accepting that, for the foreseeable future, only semi-empirical potential energy surfaces will be available for estimating reaction rate constants of many interesting molecules, this study begins to explore methods for validating such surfaces. The example chosen is that of a very simple reaction, for which it has long been known that standard classical trajectory methods yield grossly incorrect rate constants. Only two facets of the problem are addressed, whether energy can flow freely between different parts of the molecule, and the energy profile of those molecules reacting relative to the whole energy distribution profile.Despite the simplicity of the two avenues explored here, some aspects of our understanding have been improved, and a number of new ones have been observed, as follows:
(a) It is shown that the proposed heuristic measure, ϕc, of the degree of chaos behaves appropriately as the total chaos limit is approached – Fig. 1(b).
(b) With an improvement in the earlier13 calculation of ϕc, the scatter of values for CH3NC is reduced, and the resulting degree of chaos rises almost linearly with increasing energy; however, the corresponding curve for CH3CN tends to rise much faster at higher energy and may be approaching the 100% limit – Fig. 2.
(c) For a given energy, there is no correlation between the lifetime and the degree of chaos, ϕc.
(d) The fact that all trajectories eventually react means that this particular PE surface does not impose strict “intrinsic non-RRKM behaviour” but a modified version of it.
(e) Assuming the approximate equivalence of ϕc with the fch of Baer and Hase,17 scaling the computed lifetimes with the inverse of ϕc yields rate coefficients fairly close to the experimentally derived values – Fig. 3.
(f) It also appears to resolve the problem with microscopic reversibility.
(g) A deliberate attempt to amplify the degree of computational chaos by periodic reversal of momenta achieves approximately the same result – Fig. 3.
(h) Application of the same reversal procedure to CH3CN trajectories causes little or no acceleration at high energies, consistent with the high-energy behaviour of ϕc in Fig. 2, meaning that the reverse reaction rate is close to the required value; this implies that reaction impediment is very much weaker on the CH3CN side of the potential barrier than it is for the forward reaction.
(i) For all total energy values studied, and for both CH3NC and CH3CN, the distribution of potential energy followed a sinm shape, with m ∼ 4.3; likewise, the potential energy of reacting molecules also followed a sinm shape, but with m ∼ 12 – Fig. 4.
(j) The fact that m ∼ 12 for both forward and reverse reactions proves that microscopic reversibility must hold if access to the transitional configurations is equal on both sides of the barrier.
(k) Inclusion of momentum reversals every 0.1 ps at 7.0 eV above the global minimum moves the sinm curves downward by about 0.5 eV but does not change the values of m; hence, computational chaos does not affect dynamical chaos – Fig. 5 and discussion thereof.
(l) Downward movement of the mean potential energy towards the saddle point accelerates the reaction: hence the defect(s) in the PE surface must be in the mid-energy range, but mainly on the forward side.
(m) Choice of random intervals between the momentum reversals shows that no spurious spectral features arise, but that the longer the average interval, the longer the average lifetime.
Discussion
It is shown decisively that for the test reaction, although the potential energy surface was carefully crafted, there is a strong impediment to the flow of energy for CH3NC molecules to access the transition state, but a much weaker one for the CH3CN molecule to do so. Once these impediments are removed, the equivalence of the reactant energy profiles for forward and reverse processes guarantees that microscopic reversibility will ensue. This will be possible once an ab initio potential energy surface becomes available, by showing which volumes of configuration space are being excluded; recent machine-learning computer methods29 could be invaluable in the remediation of defects in this PE surface.The adjustments needed in this case could also give clues for the improvement of other similar empirical surfaces where ab initio data are not available, for example the dissociation of the peroxy radical CH3–O–O–N–O → CH3O + NO2 studied by Stimac and Barker:30 they also found the rate a factor of 20 too small but, as well, the ensemble decay resembled that of a triatomic molecule.31 Of course, this factor of 20 could just be a coincidence as the potential energy surface is far more tangled, and there might be significant complications from “roaming” effects, such are found in the dissociation of other nitro-compounds.32 We should note that for a dissociation reaction, the comparison of forward and reverse reactions is more difficult than for a simple isomerisation process.33
As already suggested, the scaling of computed k(E) values with 1/ϕc appears attractive, but it is only by comparison with results when using a true ab initio potential surface that it can be shown to be valid. On the other hand, the use of reversals to impose massive amounts of computational chaos, artificially planting trajectories on inaccessible reaction paths seems to provide a crude, but extremely powerful, qualitative mechanism for detecting bottlenecks in the activation process. How it distorts the potential energy profiles in Fig. 5, and thereby reduces the trajectory lifetimes is very puzzling; however, this reversal procedure does not represent any real physical process, so the results may remain as an intellectual curiosity for now.
Possibly more instructive are the two kinds of potential energy plots. For the overall PE distribution, it is a simple matter to save the values of the energy at a large set of times for any non-reacting trajectory and sort the values into bins. However, for the reactive PE profiles, a very minimum of 2000 trajectories is needed to generate a useful histogram. These profiles have three important uses: (i) the forward and reverse profiles must agree; (ii) the existence of only one simple sinm feature may help to guard against an unexpected alternative reaction channel; and (iii) that the values of m for the overall and the reactive profiles should not differ wildly, as the do here. Item (iii) is a matter of speculation, based on the observation of the similarity of the two values of m found in the only known calculation of this kind using an ab initio potential energy surface.11
Extension to larger molecules adds some complications. In addition to the increased complexity of the potential function, the number of inter-atomic distances needed to calculate (t) increases rapidly, e.g. for a 10-atom system there are 45 distances, but present results suggest that only nearest and next-nearest neighbour interactions make any significant contribution to the spectrum itself, or to ϕc; however there could be large but compact molecules, like neopentane or di-tert-butyl peroxide, where this may not be the case.
Finally, the zero-point energy problem has to be addressed. During the period 1989–1999, many ideas were discussed, falling roughly into two classes, the reversal of trajectories as they tried to enter the forbidden region,28,34–37 or ideas related to the fraction of time trajectories spend traversing the region below the zero-point level.10–12,38,39 Recently, the first approach has been exploited beautifully to describe the internal motions of small water clusters.40,41 However, this degree of sophistication for large organic molecules, projecting the motions into normal modes could be prohibitive, but a simple trial-and-error momentum reversal of one or a few atomic motions to prevent crossing below the zpe could probably be found.
The second approach has been shown to be exact in the RRKM limit,39 and the time fraction can be followed easily during the trajectory calculation, estimated by state-counting methods,10,11 or simply from the potential energy distributions as shown in Fig. 4.
In either case, if it is thought that tunnelling may be an issue, trajectories may penetrate the potential barrier below its maximum, and protocols for decay of the associated wave packet inside the classically forbidden barrier region need to be established.10
To summarise: momentum reversals are a useful tool for detecting some energy redistribution flaws in potential energy surfaces, but it is unclear how this works; attempts to quantify the degree of chaos show some interesting aspects but, for now, the results are tentative; potential energy distributions, as shown in Fig. 4, are especially informative for isomerisation reactions.
References
- E. C. Kemble, The Fundamental Principles of Quantum Mechanics, Dover, New York, 1937, p. 376 Search PubMed.
- L. S. Kassel, Kinetics of Homogeneous Gas Reactions, Chemical Catalog Co., New York, 1932 Search PubMed.
- K. A. Holbrook, M. J. Pilling and S. H. Robertson, Unimolecular Reactions, Wiley, New York, 2nd edn, 1996 Search PubMed.
- F. W. Schneider and B. S. Rabinovitch, J. Am. Chem. Soc., 1962, 84, 4215–4230 CrossRef CAS; F. W. Schneider and B. S. Rabinovitch, J. Am. Chem. Soc., 1963, 85, 2365–2370 CrossRef.
- C. K. Yip and H. O. Pritchard, Can. J. Chem., 1970, 48, 2942–2944 CrossRef CAS.
- H. O. Pritchard, R. G. Sowden and A. F. Trotman-Dickenson, Proc. R. Soc. London, Ser. A, 1953, 217, 563–571 CrossRef CAS.
- D. H. Shaw and H. O. Pritchard, Can. J. Chem., 1968, 46, 2721–2724 CrossRef CAS.
- J. M. Bowman, B. J. Braams, S. Carter, C. Chen, G. Czakó, B. Fu, X. Huang, E. Kamarchik, A. R. Sharma, B. C. Shepler, Y. Wang and Z. Xie, J. Phys. Chem. Lett., 2010, 1, 1866–1874 CrossRef CAS.
- B. G. Sumpter and D. L. Thompson, J. Chem. Phys., 1987, 87, 5809–5819 CrossRef CAS PubMed.
- D. Shen and H. O. Pritchard, J. Chem. Soc., Faraday Trans., 1996, 92, 1297–1304 RSC.
- H. O. Pritchard, S. R. Vatsya and D. Shen, J. Chem. Phys., 1999, 110, 9384–9389 CrossRef CAS PubMed.
- H. O. Pritchard, Cent. Eur. J. Chem., 2011, 9, 753–760 CrossRef.
- H. O. Pritchard, RSC Adv., 2013, 3, 18550–18556 RSC.
- S. Glasstone, K. J. Laidler and H. Eyring, The Theory of Rate Processes, McGraw-Hill, New York, 1941, pp. 98–99, 184–193 Search PubMed.
- R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics, Oxford University Press, Oxford, 1974, p. 164 Search PubMed.
- J. L. Collister and H. O. Pritchard, Can. J. Chem., 1976, 54, 2380–2384 CrossRef CAS.
- T. Baer and W. L. Hase, Unimolecular Reaction Dynamics, Theory and Experiments, Oxford University Press, Oxford, 1996, pp. 305–307 Search PubMed.
- W. H. Press, B. P. Flannery, S. A. Teukolsky and W. T. Vetterling, Numerical Recipes: The Art of Scientific Computing, Cambridge University Press, Cambridge, 1985 Search PubMed.
- T. Shimanouchi, Tables of Molecular Vibrational Frequencies, National Bureau of Standards, Washington, D.C., NSRDS-NBS-39, 1972, p. 85 Search PubMed.
- I. Melbourne and G. A. Gottwald, Nonlinearity, 2008, 21, 179–189 CrossRef.
- D. Shen and H. O. Pritchard, Int. J. Chem. Kinet., 1994, 26, 729–736 CrossRef CAS PubMed.
- T. D. Sewell and D. L. Thompson, J. Chem. Phys., 1990, 93, 4077–4093 CrossRef CAS PubMed.
- T. D. Sewell and D. L. Thompson, J. Chem. Phys., 1992, 96, 8006–8022 CrossRef CAS.
- D. L. Bunker and W. L. Hase, J. Chem. Phys., 1973, 59, 4621–4632 CrossRef CAS PubMed.
- W. Forst, J. Phys. Chem., 1972, 76, 342–348 CrossRef CAS.
- W. L. Hase and D. G. Buckowski, J. Comput. Chem., 1982, 3, 335–343 CrossRef CAS PubMed.
- D. Shen and H. O. Pritchard, J. Chem. Soc., Faraday Trans., 1996, 92, 4357–4360 RSC.
- G. H. Peslherbe and W. L. Hase, J. Chem. Phys., 1994, 100, 1179–1189 CrossRef CAS PubMed.
- P. Gasparotto and M. Ceriotti, J. Chem. Phys., 2014, 141, 174110 CrossRef PubMed.
- P. J. Stimac and J. R. Barker, J. Phys. Chem. A, 2008, 112, 2553–2562 CrossRef CAS PubMed.
- W.-T. Chan, D. Shen and H. O. Pritchard, J. Chem. Soc., Faraday Trans., 1995, 91, 1717–1721 RSC.
- J. M. Bowman, Mol. Phys., 2014, 112, 2516–2528 CrossRef CAS PubMed.
- N. J. B. Green and S. H. Robertson, Chem. Phys. Lett., 2014, 605–606, 44–46 CrossRef CAS PubMed.
- J. M. Bowman, B. Gazdy and Q. Sun, J. Chem. Phys., 1989, 91, 2859–2862 CrossRef CAS PubMed.
- W. H. Miller, W. L. Hase and C. L. Darling, J. Chem. Phys., 1990, 91, 2663–2668 Search PubMed.
- A. J. C. Varandas and J. M. C. Marques, J. Chem. Phys., 1994, 100, 1908–1920 CrossRef CAS PubMed.
- Y. Guo, D. L. Thompson and T. D. Sewell, J. Chem. Phys., 1996, 104, 576–582 CrossRef CAS PubMed.
- D. Shen and H. O. Pritchard, J. Chem. Soc., Faraday Trans., 1995, 91, 3747–3750 RSC.
- K. F. Lim, J. Chem. Soc., Faraday Trans., 1997, 93, 669–672 RSC.
- G. Czakó, A. L. Kaledin and J. M. Bowman, J. Chem. Phys., 2010, 132, 164103 CrossRef PubMed.
- G. Czakó, A. L. Kaledin and J. M. Bowman, Chem. Phys. Lett., 2010, 500, 217–222 CrossRef PubMed.
Footnotes |
| † In the original proposal,13 distances were tabulated at 5 fs intervals; results for 2.5 fs intervals show less scatter, but the use of 1 fs intervals shows little further improvement. |
| ‡ This probably arises because the new reversal process is programmed with fewer instructions, thereby minimising the growth of computational chaos; the new results are preferable as the older ones place the low-energy rate well above the experimental estimate. |
| This journal is © The Royal Society of Chemistry 2015 |