
Theoretical studies on the photoisomerization mechanism of osmium(II) sulfoxide complexes†
Huifang Li*a,
Lisheng Zhanga,
Yanfei Wanga and
Xiaolin Fan*ab
aKey Laboratory of Organo-Pharmaceutical Chemistry, Gannan Normal University, Ganzhou 341000, P. R. China. E-mail: huifang0801@gmail.com; fanxl@gnnu.edu.cn
bMaterial and Chemical Engineering Department, Pingxiang University, Pingxiang 337055, P. R. China
First published on 30th June 2015
Abstract
Photochromic compounds, employing photonic energy for excited-state bond rupture and bond construction processes, are excellent candidates for application as optical molecular information storage devices. In this work, a dispersion corrected density functional theory (DFT-D) study was carried out on the photoisomerization mechanism of the osmium sulfoxide complex, [Os(bpy)2(DMSO)2]2+ (bpy = 2,2′-bipyridine; DMSO = dimethyl sulfoxide), which features a pronounced photochromic character based on an ultrafast photo-triggered linkage isomerization located at the SO-ligand. Calculated results demonstrate that the Os–S → Os–O isomerization proceeds adiabatically on the potential energy surface (PES) of the lowest metal-to-ligand charge transfer excited states 3MLCTS → 3MLCTO, and the metal centered ligand field (3LFS) excited state is not involved. However, attributed to the weakened Os–DMSO bond-strength upon Os–S to Os–O isomerization, the population of the 3LFO excited state is thermally accessible. Therefore, photodissociation of DMSO from the Os center occurs via homolytic cleavage of the Os–O bond after an avoided curve crossing between the lowest 3MLCTO state and the 3LFO repulsive state along the stretching coordinate of the Os–O bond.
1. Introduction
Due to their excited-state bond rupture and bond construction characters, photochromic complexes based on d6 transition metal complexes are promising and versatile candidates for applications in light energy storage and light-activated switches.1–6 Among them, polypyridine ruthenium(II)7–10 and osmium(II)11,12 sulfoxide complexes, in which the intramolecular isomerization of the coordinated sulfoxide group from the Mn+–S to Mn+–O linkage mode can be triggered by external light signal, are highly appealing and have attracted a great deal of attention.Intramolecular isomerization of the sulfoxide group in polypyridine ruthenium(II) complexes was firstly observed for [Ru(bpy)2(DMSO)2]2+ (bpy = 2,2′-bipyridine; DMSO = dimethyl sulfoxide) by the group of F. R. Keene.13 In their studies, a color change from yellow to red was observed due to the photo-induced one DMSO ligand isomerization upon exposure to sunlight or UV irradiation. Soon afterwards, photochromic character of osmium sulfoxide complexes was reported by the group of J. J. Rack.14 In their studies, photo-induced one DMSO ligand isomerization was observed in [Os(bpy)2(DMSO)2]2+, as displayed in Fig. 1. Correspondingly, their absorption maximum was shifted from 355 nm to a new peak 403 nm by such molecular rearrangement. In the last few years, many investigations have been devoted to the synthesis and characterization studies for such polypyridine ruthenium or osmium complexes with photoisomerizable sulfoxide group.15–22 Except for experimental achievements, theoretical studies have also been carried out for exploring the electronic and photophysical properties of such transition-metal based chromophores.23–27 It is found that the HOMO–LUMO energy gap is decreased by Mn+–S → Mn+–O linkage isomerization due to the decreased electron transfer amount from surrounding ligands to central Mn+, which makes the absorption of such photochromic ruthenium(II) or osmium(II) sulfoxide complexes red shifted.26
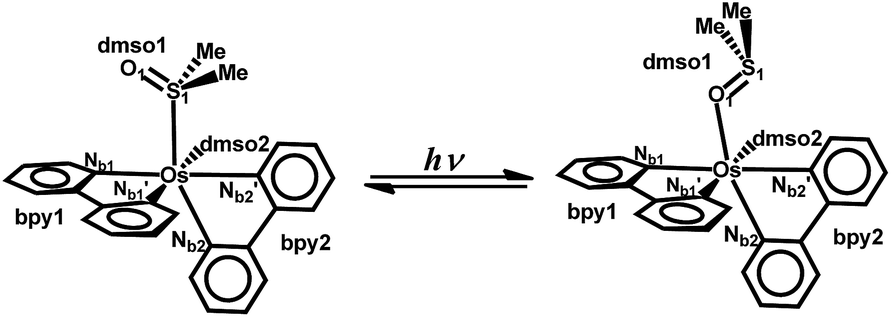 | ||
| Fig. 1 Chemical structures of the S-bonded (left) and O-bonded (right) isomers of [Os(bpy)2(DMSO)2]2+ complexes with the labeling scheme used throughout this work. | ||
It has been realized that quite different excited-state characters can be formed after one-electron excitation from the ground state (1GS) attributed to the different orbital parentage.28 The potential energies of the ground state and two triplet states are displayed in the model diagram, Fig. 2. The two triplet states are the metal-to-ligand charge transfer (3MLCT) and the metal centered excited ligand field (3LF) excited state. Among them, 3MLCT indicates the lowest excited triplet state of transition-metal complexes formed by promotion of an electron from a πM metal orbital to the πL* ligand orbitals, which exhibits long lifetime and intense luminescence emission. 3LF indicates a higher triplet energy involving the bonding and antibonding orbitals of a Mn+–L bond between surrounding ligand and Mn+ center, which often leads to fast radiationless decay to the ground state/or ligand dissociation reactions. According to previous report, 3MLCT excited state will be raised inevitably to a region very close to or even higher than the upper lying 3LF excited state by the dissociation along the Mn+–L bond.28 Then, these two states mix and intersect, which is called avoided crossing. If the energy barrier (Ea) from left to right can be overcome, 3LF becomes the lowest triplet state in the stretched Mn+–L bond region instead of 3MLCT in the equilibrium geometry region on the left of avoided crossing. As a result, the transition-metal complexes will undergo fast homolytic dissociation following the repulsive 3LF curve. It has been demonstrated that 3LF excited states play a central role in the photoisomerization mechanism of photochromic polypyridine ruthenium sulfoxide complexes.27 Thereby, nonadiabatic pathways for such photoisomerization process in photochromic [Ru(bpy)2(OSO)]+ can be explained due to their fast radiationless decay to the ground state from the 3LF states.8
 | ||
| Fig. 2 Model potential–energy curves of the ground state and two relevant excited triplet states of transition metal (Mn+) complexes along the stretching of one Mn+–L bond between surrounding ligand and Mn+ center (see text for a detailed description). | ||
However, it is still unclear that whether the 3LF excited state is involved in the photoisomerization mechanism of osmium sulfoxide complexes or not. For better understanding the extra-light triggered intramolecular isomerization mechanism of osmium sulfoxide complexes, intrinsic reaction paths (IRPs) for the Os–S1 → Os–O1 and Os–O1 → Os–S1 processes in ground and excited states of [Os(bpy)2(DMSO)2]2+ complex were examined theoretically.
2. Computational details
Considering Os–S and Os–O bonds are weak interactions and the bond lengths are overestimated by current density functional theory, the empirical dispersion correction29 was added to the density functional theory (DFT) with Becke's and Johnson's rational damping function30 in optimization calculations, and dubbed this variant DFT-D3(BJ). Based on the results from a number of benchmark calculations, collected in Table S1,† the appropriate PBE0-D3(BJ)31 exchange correlation functional was employed for the fully gradient optimization of the osmium complexes. Two basis set (BS1 and BS2) was considered. BS1 is made of SDD32 for osmium, a correlation-consistent polarized double-ζ basis set (cc-pVDZ)33 for H atoms, and a correlation-consistent polarized triple-ζ basis set (cc-pVTZ)33 for C, N, O and S atoms. BS2 is made of pseudopotential-based augmented correlation-consistent basis set (aug-cc-pVDZ-pp)34 for osmium and aug-cc-pVDZ for non-metal atoms. In addition, conductor-like polarizable continuum model (CPCM)35 using solvent acetonitrile (ε = 35.688) was also considered for optimization calculations of the involved geometries. Harmonic vibrational frequencies were calculated analytically at the same level to ascertain the nature of the optimized structures. The QST3 algorithm was employed for the location of the transition structures (TS) which were confirmed by having only one imaginary frequency with the corresponding eigenvector pointing toward the reactants and products. In addition, intrinsic reaction coordinate (IRC)36,37 calculations were also performed to make sure that the optimized transition states were connected with two relevant minima.To shed light on the nature of the excited states of the S- and O-linked osmium complexes, vertical transition energies were calculated on the basis of the optimized S0 and T1 structures via time-dependent DFT (TD-DFT)38 at the same level (BS1) which promises a way forward to study excited states of large metallic complexes. Natural transition orbital (NTO)39 analyses were performed to examine the nature of the excited states.
Charge distribution was evaluated with the natural population analysis (NPA)40 for the aim to examine the degree of charge transfer between surrounding ligands and central osmium.
All calculations were performed by using the Gaussian 09 package.41
3. Results and discussion
Photo-induced one DMSO ligand isomerization upon exposure to sunlight or UV irradiation has been observed experimentally in polypyridine osmium(II) complex, [Os(bpy)2(DMSO)2]2+.14 Chemical structures of these two different isomers with S1- and O1-linkage mode are displayed in Fig. 1. Optimized geometrical parameters are collected in Table 1, available experimental results14 are also included for comparison. A good agreement between them can be obtained. NPA charge distribution results for these isomers in the ground and excited states are listed in Table 2. Calculated reaction energies as well as energy barriers for the intramolecular bond rupture and bond construction processes in [Os(bpy)2(DMSO)2]2+ are given in Table 3. It is found that the influences of the electron density redistribution induced by one-electron excitation on the intrinsic reaction paths (IRPs) of intramolecular isomerization of [Os(bpy)2(DMSO)2]2+ are significantly.| Bond | S-bonded | O-bonded | |||||
|---|---|---|---|---|---|---|---|
| 1GSS | Exptl.a | 3MLCTS | 3LFS | 1GSO | 3MLCTO | 3LFO | |
| a Ref. 14. | |||||||
| Os–S1 | 2.286 | 2.276 | 2.381 | 2.832 | — | — | — |
| Os–O1 | — | — | — | — | 2.147 | 2.058 | 2.830 |
| S1–O1 | 1.494 | 1.471 | 1.481 | 1.512 | 1.551 | 1.574 | 1.521 |
| Os–S2 | 2.286 | 2.268 | 2.342 | 2.304 | 2.271 | 2.345 | 2.292 |
| S2–O2 | 1.494 | 1.477 | 1.483 | 1.494 | 1.491 | 1.482 | 1.492 |
| Os–Nb1 | 2.009 | 2.100 | 2.118 | 2.131 | 2.067 | 2.083 | 2.145 |
| Os–N′b1 | 2.101 | 2.091 | 2.101 | 2.246 | 2.038 | 2.052 | 2.268 |
| Os–Nb2 | 2.101 | 2.097 | 2.076 | 2.091 | 2.088 | 2.078 | 2.089 |
| Os–N′b2 | 2.009 | 2.094 | 2.024 | 2.071 | 2.094 | 2.086 | 2.059 |
| α-(O1–S1–Os)/α-(S1–O1–Os) | 116.0 | 115.8 | 113.5 | 77.0 | 120.6 | 123.6 | 116.8 |
| α-(S1–Os–Nb1)/(O1–Os–Nb1) | 98.0 | 97.8 | 99.6 | 95.6 | 91.4 | 91.1 | 83.8 |
| α-(O2–S2–Os) | 116.3 | 116.0 | 115.2 | 118.1 | 117.9 | 118.4 | 117.9 |
| S-bonded | O-bonded | |||||
|---|---|---|---|---|---|---|
| 1GSS | 3MLCTS | 3LFS | 1GSO | 3MLCTO | 3LFO | |
| a Q (ligand) indicates the charge on all the atoms in ligand. | ||||||
| S1 | 1.594 | 1.568 | 1.204 | 1.228 | 1.218 | 1.218 |
| O1 | −0.952 | −0.931 | −1.007 | −0.856 | −0.818 | −0.991 |
| Os | −0.443 | −0.049 | 0.353 | −0.062 | 0.351 | 0.355 |
| DMSO1 | 0.569 | 0.634 | 0.064 | 0.330 | 0.407 | 0.121 |
| DMSO2 | 0.569 | 0.577 | 0.479 | 0.519 | 0.592 | 0.467 |
| BPY1 | 0.652 | 0.696 | 0.523 | 0.619 | 0.018 | 0.497 |
| BPY2 | 0.652 | 0.142 | 0.581 | 0.594 | 0.631 | 0.560 |
| BS1 | BS2 | |||
|---|---|---|---|---|
| Ea | Er | Ea | Er | |
| 1GSS → 1GSO | 1.29 | 0.27 | 1.28 | 0.20 |
| 3MLCTS → 3MLCTO | 0.45 | −0.27 | 0.36 | −0.19 |
| 1GSO → 1GSS | 1.01 | −0.27 | 1.08 | −0.20 |
| 3MLCTO → 3LFO | 0.56 | 0.42 | 0.41 | 0.28 |
3.1. Os–sulfoxide bond-strength changes upon one-electron excitation
As listed in Table 1, optimized Os–S1 bond in the ground state (1GSS) of S1-[Os(bpy)2(DMSO)2]2+ is 2.286 Å, which is shorter than that in the metal-to-ligand charge transfer excited state (3MLCTS) by about 0.095 Å. On the contrary, the Os–O1 bond in the 1GSO state is longer than that in the 3MLCTO state for about 0.089 Å. It means the Os–S1 bond is weakened, while Os–O1 bond is strengthened upon one-electron excitation.Frontier molecular orbitals (MOs) play an important role in photoresponsive materials because location of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is quite reasonable for the description of the first excited singlet transition. As shown in Fig. 3, for these [Os(bpy)2(DMSO)2]2+ isomers, the HOMO is mainly distributed on central osmium, while the LUMO is mainly distributed on the bpy ligands. Thereby, the 3MLCTO and 3MLCTO excited states were formed with one-electron promotion from a πOs metal orbital to the πbpy* bpy ligand orbitals. Electron configuration and singly occupied molecular orbitals of the excited states are shown in Fig. 4. NPA charge calculation results demonstrate that the charge distributed on central osmium is decreased by 0.394 and 0.413 e, respectively, upon one-electron excitation from S1- and O1-linked isomers.
 | ||
| Fig. 3 Chemical structures of the S-bonded (left) and O-bonded (right) isomers of [Os(bpy)2(DMSO)2]2+ complexes with the labeling scheme used throughout this work. | ||
 | ||
| Fig. 4 Electron configuration for the singly occupied molecular orbitals of the excited states of [Os(bpy)2(DMSO)2]2+. | ||
As known, S-bonding is favored in the case of ‘soft’ metal atoms where the sulfoxides act as moderate π-acceptor ligands,42 so that the metal to sulfur bond is strengthened when electron distribution on central metal is increased. Thereby, weakened Os–S1 bond upon one-electron excitation from a πOs orbital to the πbpy* orbitals can be explained. Differently, it has been proved that O-bonding is favored in the case of ‘hard’ metal atoms where the sulfoxides act as moderate σ-donor ligands, so that the metal–oxygen bond is strengthened when electron distribution on central metal is decreased. Thereby, the Os–O1 bond is strengthened by one-electron excitation from a πOs orbital to the πbpy* orbitals.
The variation trends of the S–O bond distances are also rationalized considering the metal–sulfoxide bonding changes upon one-electron excitation. As listed in Table 2, the weakened Os–S1 bond-strength reduces the positive charge of sulfur atom from 1.594 to 1.568 e, and thus the electronegativity of S1 atom is decreased. Therefore, there is additional electron transfer from O1 to S1 (the charge on O1 is decreased from −0.952 to −0.931 e) for compensation, which leads to the increased double bond character of the S1–O1. As a result, the S1–O1 bond in the ground state of S1-[Os(bpy)2(DMSO)2]2+ (1GSS) is decreased from 1.494 to 1.481 Å upon one-electron excitation. For the O1-linked isomer, the charge on O1 atom is changed from −0.856 to −0.818 e by one-electron excitation, which makes the electronegativity of O1 atom increased. Due to the equalization of atom electronegativities,43 this is compensated by electron transfer from the sulfur atom and the methyl groups which makes the positive charge on the S1 atom decreased from 1.228 to 1.218 e. Furthermore, attributed to the increased O1 electronegativity, the π-electron transfer from O1 to S1 is decreased. As a result, the S–O bond-strength is weakened and its bond-length is increased from 1.551 to 1.574 Å.
3.2. Intrinsic reaction paths
As shown in Fig. 5, S1-[Os(bpy)2(DMSO)2]2+ is stable than its O1-linked isomer, O1-[Os(bpy)2(DMSO)2]2+, for about 0.20 eV because Os–S linkage mode of DMSO is preferred in the ground state of Os(II) complex. Energy barrier for the isomerization from S- to O-linkage is about 1.08 eV. Computational results demonstrated that the energy barrier between S1- and O1-linked isomers in the lowest excited states is lower than that in the ground states by about 0.92 eV. It means such intramolecular isomerization process should be much easier to proceed in the lowest excited state. Thereby, stimulated by external light, one DMSO ligand isomerization from Os–S1 to Os–O1 linkage mode in [Os(bpy)2(DMSO)2]2+ was observed in experimental studies. As mentioned above, the Os–S1 bond is weakened and the Os–O1 bond is strengthened by electron density redistribution upon one-electron excitation, which makes the energy of S1-[Os(bpy)2(DMSO)2]2+ higher than O1-[Os(bpy)2(DMSO)2]2+ by about 0.36 eV in the lowest 3MLCT excited states. As a result, the Os–S1 to Os–O1 isomerization process is changed from endothermic to exothermic reaction upon one-electron excitation. However, calculated results show that the S1-[Os(bpy)2(DMSO)2]2+ in the lowest 3MLCT excited state is stable than that in the 3LFS state for about 0.16 eV. This means 3MLCTS → 3MLCTO isomerization is more thermodynamically favored in the excited state compared with the 3MLCTS → 3LFS pathways. | ||
| Fig. 5 Calculated Reaction pathways for the Os–S1 to Os–O1 isomerization of [Os(bpy)2(DMSO)2]2+. Energies are in eV. | ||
It is obvious that the photoisomerization mechanism of ruthenium sulfoxide complex, [Ru(bpy)2(OSO)]+, is different with that of [Os(bpy)2(DMSO)2]2+. As reported by A. J. Göttle and his coworkers, 3LF state plays a central role in the Ru–S → Ru–O nonadiabatic photoisomerization mechanism of [Ru(bpy)2(OSO)]+ complexes.27 Thereby, no O-bonded excited state, 3MLCTO, of [Ru(bpy)2(OSO)]+ complex can be observed experimentally.8 It is proposed that the 3LFS excited state is more stable than the 3MLCTO state. Thereby, the 3LFS excited state in [Ru(bpy)2(OSO)]+ should be thermally accessible from the initial radiative 3MLCTS electron state and no 3MLCTO excited state of [Ru(bpy)2(OSO)]+ complex was observed.
Differently, along with the Os–O bond rupture in the lowest 3MLCTO excited state of O1-[Os(bpy)2(DMSO)2]2+, instead of Os–S bond construction, a new O1-linked osmium complex in the 3LFO state is formed. Optimized geometrical parameters of the O1-[Os(bpy)2(DMSO)2]2+ in the 3LFO state are listed in Table 1. It means, for the excited-state Os–O bond breaking process, nonradiative 3LFO excited state should be accessible from the initial radiative 3MLCTO electron state. As shown in Fig. 6, it is because the energy barrier for the 3MLCTO → 3LFO process (0.41 eV) is smaller than that for the 3MLCTO → 3MLCTS pathway (0.56 eV). Considering promotion to the 3LF state often leads to fast radiationless decay to the ground state or ligand dissociation reactions,44 it can be predicted that photodissociation of DMSO from Os center may occur via homolytic cleavage of Os–O bond after an avoided curve crossing between the lowest 3MLCTO state and a 3LF repulsive state along the stretching mode of the Os–O bond.
 | ||
| Fig. 6 Calculated Reaction pathways for the Os–O1 bond breaking process in [Os(bpy)2(DMSO)2]2+. Energies are in eV. | ||
This can be confirmed with TD-DFT calculation results. As shown in the Fig. 7 and Table S2,† NTO results demonstrated that the first transition (S1) of S-[Os(bpy)2(DMSO)2]2+ is from the ground state (1GSS) to 1MLCT (formed with πL* ligand orbitals and dxy, dyz or dxz metal orbitals) and the third transition (S3) is from 1GSS to 1LFS (formed with πL*ligand orbitals and dx2−y2 or dz2 metal orbitals). For O-[Os(bpy)2(DMSO)2]2+, the S1 is from 1GSO to 1MLCTO and the S2 is from 1GSO to 1LFO. The energy gap between 1MLCTS and 1LFS is lower than that between 1MLCTO and 1LFO (0.28 eV). This is in good agreement with DFT calculation results. More importantly, from TD-DFT results, it is observed that the 1MLCTO excited state is more stable that the 1MLCTS excited state. Moreover, the energy of the 1LFO excited state is lower than that of the 1MLCTS excited state. This can be used for better understanding that why the Os–S1 → Os–O1 isomerization proceeds adiabatically on the PES of MLCTS → MLCTO and the LFS excited state is not involved. Differently, for the excited-state Os–O bond breaking process, nonradiative LFO excited state should be accessible from the initial radiative MLCTO excited state.
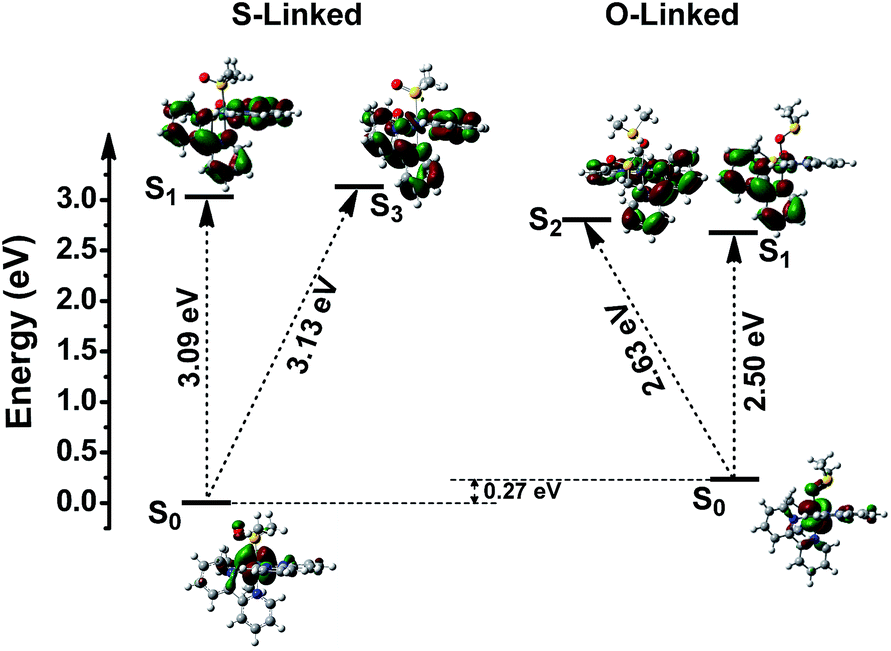 | ||
| Fig. 7 Photophysical properties of [Os(bpy)2(DMSO)2]2+ isomers obtained from TD-DFT calculations. The nature of the orbitals involved in the relevant excited states is also included. | ||
4. Conclusion
A DFT-D study was carried out on the photo-triggered intramolecular linkage isomerization of osmium sulfoxide complex, [Os(bpy)2(DMSO)2]2+, for better understanding the photoisomerization mechanism of osmium complexes as well as the role of the 3LF excited state in the bond rupture (Mn+–X) and bond construction (Mn+–Y) processes.The metal-to-ligand charge transfer excited states (3MLCTS and 3MLCTO) were formed by one-electron promotion from a πOs metal orbital to the πbpy* bpy ligand orbitals which makes the electron distribution on central osmium decreased. As a consequence, the Os–S1 bond is weakened because S-bonding is favored in the case of ‘soft’ metal atoms, while the Os–O1 bond is strengthened because O-bonding is favored in the case of ‘hard’ metal atoms. As a result, the Os–S1 to Os–O1 isomerization process is changed from endothermic to exothermic reaction upon one-electron excitation. It means such intramolecular isomerization process should be much easier to proceed in the lowest excited state.
Moreover, it is found that the Os–S1 → Os–O1 isomerization proceeds adiabatically on the PES of 3MLCTS → 3MLCTO, and the 3LFS state is not involved. Differently, along with the Os–O bond rupture in the lowest 3MLCTO excited state of O1-[Os(bpy)2(DMSO)2]2+, no Os–S bond construction is observed and a new O1-linked osmium complex in the 3LFO state is formed. It means, for the excited-state Os–O bond breaking process, nonradiative 3LFO excited state should be accessible from the initial radiative 3MLCTO electron state. As a result, photodecomposition of the DMSO ligand from the 3LFO excited state complex may occur along with the Os–O bond stretching mode because promotion to the 3LF state often leads to fast radiationless decay to the ground state or ligand dissociation reactions.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21403037, 51478123, 21463003), the Natural Science Foundation of Jiangxi Province (20142BAB213014), Jiangxi Provincial “Ganpo Talents 555 Projects” and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, PR China.References
- N. V. Mockus, D. Rabinovich, J. L. Petersen and J. J. Rack, Angew. Chem., Int. Ed., 2008, 47, 1458–1461 CrossRef CAS PubMed.
- M. N. Roberts, C.-J. Carling, J. K. Nagle, N. R. Branda and M. O. Wolf, J. Am. Chem. Soc., 2009, 131, 16644–16645 CrossRef CAS PubMed.
- T. Ishizuka, T. Sawaki, S. Miyazaki, M. Kawano, Y. Shiota, K. Yoshizawa, S. Fukuzumi and T. Kojima, Chem.–Eur. J., 2011, 17, 6652–6662 CrossRef CAS PubMed.
- S. Bonnet and J.-P. Collin, Chem. Soc. Rev., 2008, 37, 1207–1217 RSC.
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618–635 RSC.
- K. Garg, A. W. King and J. J. Rack, J. Am. Chem. Soc., 2014, 136, 1856–1863 CrossRef CAS PubMed.
- B. A. McClure, N. V. Mockus, D. P. Butcher, Jr, D. A. Lutterman, C. Turro, J. L. Petersen and J. J. Rack, Inorg. Chem., 2009, 48, 8084–8091 CrossRef CAS PubMed.
- B. A. McClure, E. R. Abrams and J. J. Rack, J. Am. Chem. Soc., 2010, 132, 5428–5436 CrossRef CAS PubMed.
- A. A. Rachford and J. J. Rack, J. Am. Chem. Soc., 2006, 128, 14318–14324 CrossRef CAS PubMed.
- B. A. McClure and J. J. Rack, Inorg. Chem., 2011, 50, 7586–7590 CrossRef CAS PubMed.
- J. J. Rack, Coord. Chem. Rev., 2009, 253, 78–85 CrossRef CAS PubMed.
- K. Garg, S. I. M. Paris and J. J. Rack, Eur. J. Inorg. Chem., 2013, 1142–1148 CrossRef CAS PubMed.
- M. K. Smith, J. A. Gibson, C. G. Young, J. A. Broomhead, P. C. Junk and F. R. Keene, Eur. J. Inorg. Chem., 2000, 1365–1370 CAS.
- (a) N. V. Mockus, J. L. Petersen and J. J. Rack, Inorg. Chem., 2006, 45, 8–10 CrossRef CAS PubMed; (b) B. A. McClure and J. J. Rack, Eur. J. Inorg. Chem., 2010, 3895–3904 CrossRef CAS PubMed.
- N. V. Mockus, S. Marquard and J. J. Rack, J. Photochem. Photobiol., A, 2008, 200, 39–43 CrossRef CAS PubMed.
- A. W. King, Y. Jin, J. T. Engle, C. J. Ziegler and J. J. Rack, Inorg. Chem., 2013, 52, 2086–2093 CrossRef CAS PubMed.
- A. A. Rachford, J. L. Petersen and J. J. Rack, Dalton Trans., 2007, 3245–3251 RSC.
- A. E. Phillips, J. M. Cole, T. d'Almeida and K. S. Low, Inorg. Chem., 2012, 51, 1204–1206 CrossRef CAS PubMed.
- B. A. McClure and J. J. Rack, Angew. Chem., Int. Ed., 2009, 48, 8556–8558 CrossRef CAS PubMed.
- B. L. Portera, B. A. McClurea, E. R. Abramsa, J. T. Engleb, C. J. Zieglerb and J. J. Rack, J. Photochem. Photobiol., A, 2011, 217, 341–346 CrossRef PubMed.
- A. A. Rachford, J. L. Petersen and J. J. Rack, Inorg. Chem., 2006, 45, 5953–5960 CrossRef CAS PubMed.
- D. P. Butcher, A. A. Rachford, J. L. Petersen and J. J. Rack, Inorg. Chem., 2006, 45, 9178–9180 CrossRef CAS PubMed.
- I. Ciofini, C. A. Daul and C. Adamo, J. Phys. Chem. A, 2003, 107, 11182–11190 CrossRef CAS.
- D. A. Lutterman, A. A. Rachford, J. J. Rack and C. Turro, J. Phys. Chem. A, 2009, 113, 11002–11006 CrossRef CAS PubMed.
- (a) H. F. Li, L. S. Zhang, H. Lin and X. L. Fan, RSC Adv., 2014, 4, 45635–45640 RSC; (b) H. F. Li, L. S. Zhang, H. Lin and X. L. Fan, Chem. Phys. Lett., 2014, 604, 10–14 CrossRef CAS PubMed.
- H. F. Li, L. S. Zhang, X. L. Fan and Y. Zhao, J. Phys. Chem. A, 2015, 119, 4244–4251 CrossRef CAS PubMed.
- A. J. Göttle, I. M. Dixon, F. Alary, J.-L. Heully and M. Boggio-Pasqua, J. Am. Chem. Soc., 2011, 133, 9172–9174 CrossRef PubMed.
- (a) S. Bonnet and J. P. Collin, Chem. Soc. Rev., 2008, 37, 1207–1217 RSC; (b) Q. Sun, S. Mosquera-Vazquez, L. M. L. Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, J. Am. Chem. Soc., 2013, 135, 13660–13663 CrossRef CAS PubMed; (c) J. T. Hewitt, J. J. Concepcion and N. H. Damrauer, J. Am. Chem. Soc., 2013, 135, 12500–12503 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- (a) A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed; (b) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef PubMed; (c) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS PubMed.
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- S. Fantacci, F. D. Angelis and A. Selloni, J. Am. Chem. Soc., 2003, 125, 4381–4387 CrossRef CAS PubMed.
- R. L. Martin and V. Affiliations, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- M. Calligaris, Coord. Chem. Rev., 2004, 248, 351–375 CrossRef CAS PubMed.
- (a) W. J. Mortier, Struct. Bonding, 1987, 66, 125 CrossRef CAS; (b) R. T. Sanderson, J. Chem. Educ., 1954, 31, 2 CrossRef CAS.
- (a) P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS PubMed; (b) Q. Sun, S. Mosquera-Vazquez, L. M. L. Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, J. Am. Chem. Soc., 2013, 135, 13660–13663 CrossRef CAS PubMed; (c) J. T. Hewitt, J. J. Concepcion and N. H. Damrauer, J. Am. Chem. Soc., 2013, 135, 12500–12503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Benchmark calculations. TD-DFT results. Optimized Cartesian coordinates, electronic structures, energies of all stationary points and partial optimization results. See DOI: 10.1039/c5ra06723e |
| This journal is © The Royal Society of Chemistry 2015 |