
Synthesis and self-assembly of well-defined binary graft copolymer and its use in superhydrophobic cotton fabrics preparation
Abstract
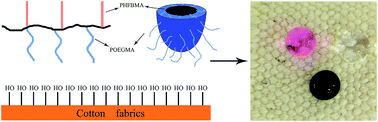
We have synthesized and characterized a series of functional binary graft copolymers PGMA-g-(PHFBMA-r-POEGMA)s(BGCs). First, PHFBMA–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, POEGMA–CCH and P(GMA-N3) were synthesized via sequential atom transfer radical polymerization (ATRP). BGCs were prepared by grafting of alkyne-end poly(hexafluorobutyl methacrylate) (PHFBMA–CCH) and poly(oligo (ethylene glycol) methyl ether methacrylate) (POEGMA–CCH) onto poly(3-azide-2-hydroxypropyl methacrylate) (P(GMA-N3)) via click chemistry. The self-assembly behaviors were investigated by combination of dynamic light scattering (DLS), transmission electron microscopy (TEM), and atomic force microscopy (AFM). Since POEGMA was soluble in water while PHFBMA was insoluble, BGCs self-assembled and produced stable PHFBMA-centered nano-micelles; then BGCs were used to fabricate hydrophobic cotton fabrics. While the PHFBMA block provided low surface free energy, the POEGMA block served as an anchor with cotton fibers, the modified cotton fabrics showed excellent superhydrophobic property. The results confirmed that fluorinated surface was formed onto substrate without changing the transparency and bulk composition of the cotton fabrics. Moreover, SEM and AFM analysis indicated that nano- and microscale roughness were created by combining BGC-based nano bumps onto surfaces of micro-sized cotton fabrics.
CH, POEGMA–CCH and P(GMA-N3) were synthesized via sequential atom transfer radical polymerization (ATRP). BGCs were prepared by grafting of alkyne-end poly(hexafluorobutyl methacrylate) (PHFBMA–CCH) and poly(oligo (ethylene glycol) methyl ether methacrylate) (POEGMA–CCH) onto poly(3-azide-2-hydroxypropyl methacrylate) (P(GMA-N3)) via click chemistry. The self-assembly behaviors were investigated by combination of dynamic light scattering (DLS), transmission electron microscopy (TEM), and atomic force microscopy (AFM). Since POEGMA was soluble in water while PHFBMA was insoluble, BGCs self-assembled and produced stable PHFBMA-centered nano-micelles; then BGCs were used to fabricate hydrophobic cotton fabrics. While the PHFBMA block provided low surface free energy, the POEGMA block served as an anchor with cotton fibers, the modified cotton fabrics showed excellent superhydrophobic property. The results confirmed that fluorinated surface was formed onto substrate without changing the transparency and bulk composition of the cotton fabrics. Moreover, SEM and AFM analysis indicated that nano- and microscale roughness were created by combining BGC-based nano bumps onto surfaces of micro-sized cotton fabrics.
 Please wait while we load your content...
Please wait while we load your content...

