
Impact of rapid ozone degradation on the structure and properties of polypropylene using a reactive extrusion process
Abstract
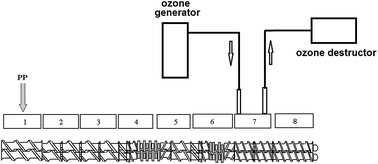
In this work, rapid ozone degradation of polypropylene (PP) was developed for the aim of rheology control using a reactive extrusion process. Experiments were carried out in a co-rotating intermeshed twin-screw extruder with varied polymer throughput and reaction temperature. Ozone was introduced into the extruder to rapidly oxidize molten PP in just several seconds period. The oxidized PP was characterized through melt flow index (MFI), rheological measurement, differential scanning calorimetry (DSC), and Fourier transform infrared (FTIR) spectroscopy tests. The influence of reactive temperature and polymer throughput on the degradation reaction was studied. It was noted that molten PP could be fast and successfully degraded during this reactive extrusion process. The oxidized PP had higher MFI than that of the origin PP resin, indicating the decrease of molecular weight of PP. Carbonyl groups were formed on the PP molecular chains. This rapid oxidization process has higher reaction efficiency than the ozone degradation of PP in solid state and no harmful byproduct would be generated from this ozonizing reaction.
 Please wait while we load your content...
Please wait while we load your content...

