
Liquid phase collagen modified graphene that induces apoptosis
Abstract
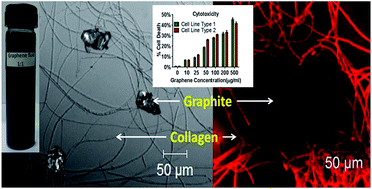
Direct exfoliation of graphite (GR) to collagen modified graphene (G) flakes using acylated collagen has been studied. The chemical structure of collagen (CL) and all liquid exfoliants studied so far have striking similarity, namely, the hydrocarbon chain length, aromatic residues and polarity. Here, CL dispersed in acetic (AA), succinic (SA) and propionic (PA) acid behaves as three different surfactants which have been used to simultaneously exfoliate and disperse nano G platelets to a colloidal form. Transmission electron microscopy micrographs show average dimensions of ∼500 × 200 nm; Moiré patterns observed at several places in all the three samples indicate periodic perturbations in the graphitic stacking and selected area electron diffraction confirms graphene formation. AFM images confirmed the same lateral dimensions with an average thickness of 1.2 nm. The Raman spectra revealed strain dependent splitting and a red shift of 2D bands, a maximum in G-PA and minimum in G-AA. X-ray photoelectron spectroscopy showed a decrease of the sp2/sp3 ratio GR post CL interaction indicating an interaction. The zeta potential, fluorescence and luminescence values changed in G, with maximum variation in G-PA; suggesting that CL dispersed in PA is the best. The bioactivity of colloidal G-PA was studied in solid, hematological and neuronal cancer cell lines. It induced reactive oxygen species and cell death in cancer cell lines and altered membrane integrity while sparing normal cells, underscoring its possible utility in cancer therapy.
 Please wait while we load your content...
Please wait while we load your content...

