
A colorimetric and turn-on fluorescent chemosensor for selective detection of Hg2+: theoretical studies and intracellular applications†
Rimi Roya,
Soumyadipta Rakshitb,
Sanjay Bhar*a and
Subhash Chandra Bhattacharya*b
aDepartment of Chemistry, Organic Chemistry Section, Jadavpur University, Kolkata 700 032, India. E-mail: sanjaybharin@yahoo.com; sanjay_bhar@chemistry.jdvu.ac.in; Fax: +91 033 24137902; Tel: +91 8697179547
bDepartment of Chemistry, Physical Chemistry Section, Jadavpur University, Kolkata 700 032, India. E-mail: sbjuchem@yahoo.com; scbhattacharyya@chemistry.jdvu.ac.in; Fax: +91 033 24146584; Tel: +91 033 24146223
First published on 31st July 2015
Abstract
A new colorimetric, “turn-on” fluorescent chemosensor (DEAS-BPH) was synthesized for selective and sensitive recognition of Hg2+ ions with no interference from environmentally relevant metal ions in a mixed organo-aqueous medium. It was found that the presence of mercury induced nearly 27-fold fluorescence enhancement. This was attributed to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal–ligand complexation, ascertained by a Job's plot, NMR titration and HRMS studies. A theoretical study was conducted to rationalize the colorimetric sensing behaviour of the probe on the basis of an intramolecular charge transfer (ICT) mechanism. In addition, morphological alteration upon complexation with mercury was explored with the aid of Field Emission Scanning Electron Microscope (FESEM) measurement. Results of MTT assay and fluorescence microscopic studies unveiled that the probe is cell permeable with low cytotoxicity. Furthermore, the reversibility of the sensor and its efficacy to function in a wide range of pH values designated the suitability to image intracellular Hg2+ ions in living HepG2 and Escherichia coli bacterial cells.
:2 metal–ligand complexation, ascertained by a Job's plot, NMR titration and HRMS studies. A theoretical study was conducted to rationalize the colorimetric sensing behaviour of the probe on the basis of an intramolecular charge transfer (ICT) mechanism. In addition, morphological alteration upon complexation with mercury was explored with the aid of Field Emission Scanning Electron Microscope (FESEM) measurement. Results of MTT assay and fluorescence microscopic studies unveiled that the probe is cell permeable with low cytotoxicity. Furthermore, the reversibility of the sensor and its efficacy to function in a wide range of pH values designated the suitability to image intracellular Hg2+ ions in living HepG2 and Escherichia coli bacterial cells.
Introduction
Mercury contamination presents a serious environmental concern throughout the world due to its high toxicity and widespread impact in the lithosphere and in water, mainly caused by its extended use in agriculture and industry.1 Both elemental and ionic mercury present in soil or in waste water are assimilated and converted by microorganisms to methylmercury, a potent neurotoxin which is consequently bioaccumulated through the food chain.2 Organomercury compounds can easily permeate the cell membranes and the blood brain barrier impairing nefrological and neurological functions. As a result, mercury exposure even at very low concentrations can cause severe metabolic, motor and cognitive disorders and sensory long term diseases in human beings.3 The extreme toxicity of mercury and its derivatives may be ascribed to its interference with thiols4 present in enzymes, proteins and DNA which disrupts the activities of the living cells and eventually leads to toxicity in humans. Hence, sensitive and selective detection of Hg2+ is currently a task of key importance rather than determination of total Hg. Quantification of mercury and its species is usually performed by conventional analytical techniques including cold-vapor atomic absorption spectrometry (CV-AAS),5 cold-vapor atomic fluorescence spectrometry (CV-AFS),6 inductively coupled plasma atomic emission spectrometry (ICP-AES),7 inductively coupled plasma mass spectrometry (ICP-MS),8 surface-enhanced Raman scattering (SERS),9 liquid chromatography (LC),10,12 gas chromatography (GC)11,12 and capillary electrophoresis (CE).12 Although these methods are unparallel in terms of sensitivity, they are associated with certain disadvantages. Time consuming, labor-intensive sample pretreatment procedures as well as costly and complicated instrumentation limit their use for fast and facile onsite analysis of mercury.Of late, the use of fluorometric sensors has emerged as one of the most promising alternatives to the conventional methods owing to their high sensitivity, selectivity and operational simplicity. Likewise, colorimetric chemosensors have been attracting a great deal of interest for “naked-eye” detection in an uncomplicated and economical manner, offering qualitative and quantitative information.13 A variety of fluorescent small-organic-molecule-based chemosensors that selectively bind to Hg2+ have been designed and developed to date. In most cases, however, the presence of mercury causes quenched fluorescence of the fluorophores because Hg2+ often acts as an efficient fluorescence quencher like many other heavy- and transition-metal (HTM) ions through an effective spin–orbit coupling.14 This phenomenon is quite disadvantageous for analytical purposes and leads to a lack of high signal output during detection. On the other hand, the sensors that exhibit fluorescence-on response upon binding with metal ions are to be favoured in practical applications over the less sensitive “turn-off” sensors due to lack of background signal, as discovered by numerous studies.15 Since chemical behaviour of Hg2+ is similar to that of Ag+, Pb2+ and Cu2+, these metals act as good competitors and different fluorescent probes end up with lack of good selectivity. Thus, the development of “turn-on” Hg2+ fluorescent sensors with high selectivity and sensitivity in living cells through a simple synthetic approach is necessary and in great demand.
N,N-Diethylaminosalicylaldehyde (DEAS) based frameworks serve as an ideal fluorophore in various fluorescence-enhanced chemosensors owing to their elongated emission wavelengths and high fluorescence quantum yield.16 In this regard, herein we reveal a new N,N-diethylaminosalicylaldehyde appended “turn-on” fluorescent, colorimetric and sulfur free, easy-to-prepare chemosensor (DEAS–BPH) for selective detection of Hg2+. The receptor is highly selective and sensitive toward Hg2+ irrespective of other interfering ions as detected by ‘naked-eye’ color change and UV-vis and emission spectral changes. The probe exhibited distinctive enhanced fluorescence response (∼27 fold) to Hg2+, which is purely attributed to an excited-state intramolecular charge transfer (ICT)17 between the donor 5-(diethylamino)-2-(iminomethyl)phenol moiety and the acceptor diphenylmethanimine units, further supported by DFT/TDDFT calculations. Thus, the probe not only provided a highly efficient sensor for recognition of Hg2+ with a low detection limit in mixed organo-aqueous solution but its low cytotoxic nature offered the possibility of using it for fluorescence imaging in living cells. Besides, to understand the role of the N,N-diethylamine moiety toward selective sensing of metal ions by ICT, a comparative study was attempted by replacing the DEAS moiety with salicylaldehyde unit (Scheme 1).
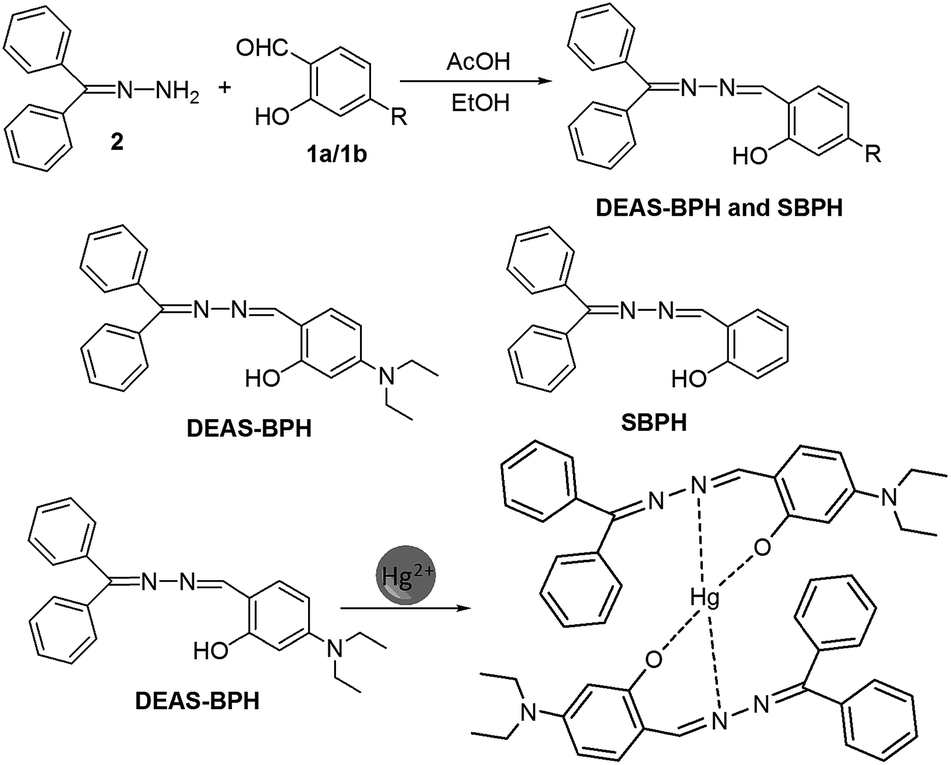 | ||
| Scheme 1 Synthetic route of DEAS–BPH and SBPH. | ||
Results and discussion
Synthesis of probe (DEAS–BPH)
As illustrated in Scheme 1, the probe DEAS–BPH was readily synthesized from the acetic acid mediated condensation reaction of 4-N,N-diethylaminosalicylaldehyde 1a with equimolecular amount of 1-(diphenylmethylene) hydrazine or benzophenone hydrazone 2 in dry ethanol for 4 h under reflux. The product was obtained as a bright yellow powder in 82% yield and it was characterized through 1H NMR, 13C NMR, FTIR and ESI-MS analysis (Fig. S1–S4†). In the FTIR spectra, the appearance of the peak at 1633 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, characteristic of imine) suggested evidence for the formation of the Schiff base. Detailed synthetic procedure of SBPH and spectroscopic analysis data are provided in the ESI.†
N, characteristic of imine) suggested evidence for the formation of the Schiff base. Detailed synthetic procedure of SBPH and spectroscopic analysis data are provided in the ESI.†
UV-vis absorption studies
Initial spectroscopic studies of the probe DEAS–BPH were executed in CH3CN–H2O (4:1 v/v) solution buffered to pH 7.2 using HEPES (2-[4-(2-hydroxyethyl)-1-piperazinyl] ethanesulfonic acid) buffer solution (5 mM). As depicted in the spectrophotometric titration curve (Fig. 1), free DEAS–BPH exhibited an absorption band at 405 nm. Upon stepwise addition of Hg2+ (0–25 μM) to the solution of the probe in CH3CN–H2O (4:1 v/v), a new band centered at 448 nm arose which became more intense with increasing concentration of Hg2+, and the color of the solution turned from pale yellow to bright yellow. The enhanced red-shifted ICT band infers that the donor unit of the receptor (–NEt2) was not involved in binding with the metal ion, therefore, Hg2+ was able to augment the ICT effect after complexing with the probe and the presence of Hg2+ could be easily monitored by the naked eye. Moreover, no discernible spectral alterations were observed in the presence of various competitive metal ions such as Na+, K+, Ag+, Mn2+, Cd2+, Cu2+, Fe3+, Al3+, Cr3+, Pb2+, Co2+, Mg2+, Zn2+ and Ni2+ (all the salts studied herein were taken as nitrates to eliminate the effect of anions, Fig. S5†), indicating that the UV-vis response of DEAS–BPH is highly Hg2+ specific.
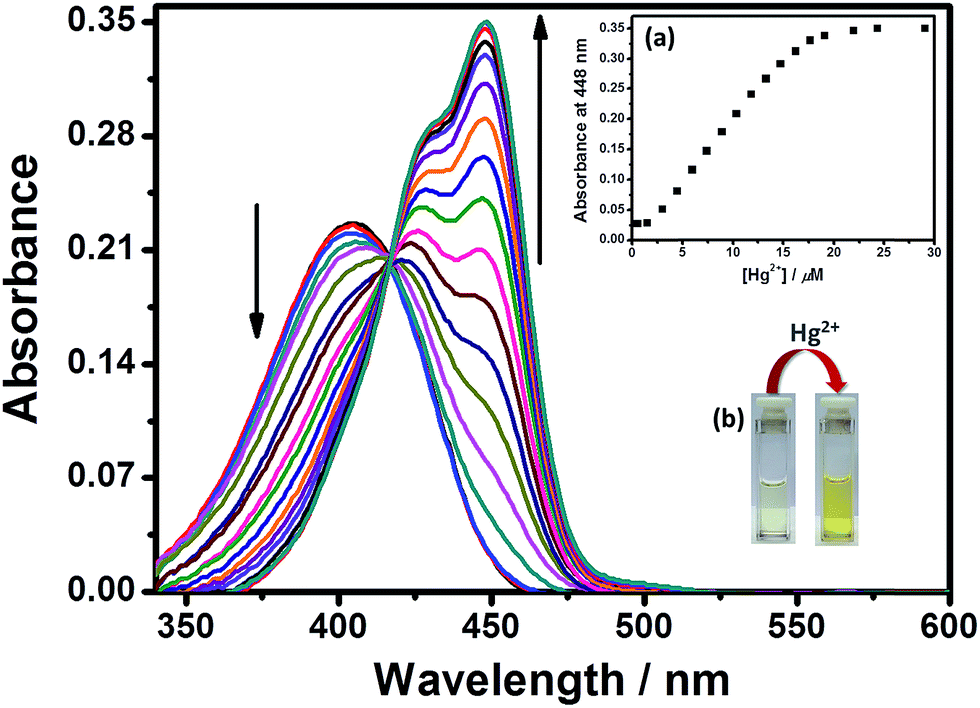 | ||
| Fig. 1 UV-vis absorption titration spectra of DEAS–BPH (6 μM) in the presence of Hg2+ (0–25 μM) at pH = 7.2 in CH3CN–H2O (4:1 v/v). Inset: (a) absorption enhancement at 448 nm as a function of Hg2+ concentration, (b) the naked eye color change of DEAS–BPH on addition of Hg2+ (4 equivalents). | ||
In this context it was interesting to note that the absorption spectrum of SBPH was remarkably different from that of DEAS–BPH. The former displayed two absorption bands at 301 and 348 nm in CH3CN–H2O (4:1 v/v) under identical condition. Upon addition of 4 equiv. of various metal ions, including Na+, K+, Ag+, Mn2+, Cd2+, Cu2+, Fe3+, Al3+, Cr3+, Pb2+, Co2+, Mg2+, Zn2+, Ni2+ and Hg2+ (Fig. S6, ESI†), no shifts in the peak positions or appearance of any new peak was observed. Hence, it was evident that the N,N-diethylamine substituent at para-position with +R effect preferentially promotes ICT in comparison to the substrate where no substituent is present at the same location.
Fluorescence studies
The fluorescence titration of the probe DEAS–BPH (6 μM) with Hg2+ has also been conducted in the same HEPES buffer (5 mM, pH 7.2) in CH3CN–H2O solution (4:1, v/v). As delineated in Fig. 2, upon addition of increasing concentrations of Hg2+ (0–20 μM) the fluorescence intensity enhanced linearly together with the maximum emission peak underwent a progressive red shift from 474 nm for free ligand to 489 nm on complexation with Hg2+. Upon excitation at the isosbestic point (λex = 416 nm) the fluorescence intensity of the complex is less than that when excited at the absorption maximum of the complex (λex = 450 nm) (Fig. S8†). However, other competitive metal ions for instance Na+, K+, Ag+, Mn2+, Ca2+, Cd2+, Cu2+, Fe3+, Al3+, Cr3+, Pb2+, Co2+, Mg2+, Zn2+ and Ni2+ (20 μM) have hardly responded to the emission of the probe when coexisted with Hg2+ (20 μM) (Fig. 3a and S9†). The fluorescence spectrum of SBPH showed an emission band at 558 nm when excited at 348 nm. In the presence of 3 equivalents of different metal ions such as Na+, K+, Ag+, Mn2+, Cd2+, Cu2+, Fe3+, Al3+, Cr3+, Pb2+, Co2+, Mg2+, Zn2+, Ni2+ and Hg2+, the emission spectra displayed neither any change nor developed any other peak (Fig. S7, ESI†). This further established that the –NEt2 group plays a significant role in promoting the ICT effect toward Hg2+ sensing.
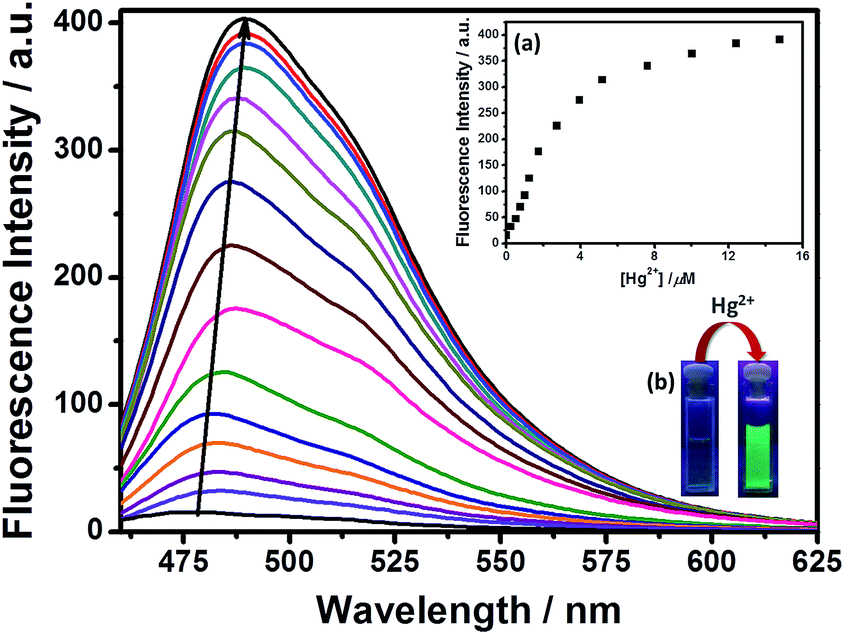 | ||
| Fig. 2 Fluorescence emission spectra of DEAS–BPH (6 μM) with gradual addition of Hg2+ (0–20 μM) at pH = 7.2 in CH3CN–H2O (4:1 v/v): λex = 450 nm, λem = 489 nm. Inset: (a) fluorescence enhancement at 489 nm as a function of Hg2+ concentration, (b) the naked eye color change of DEAS–BPH under UV-lamp (254 nm) with the addition of Hg2+ (3 equivalents). | ||
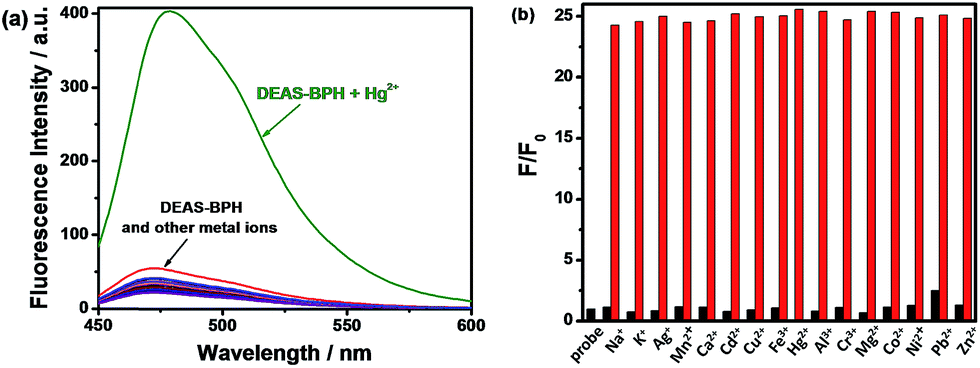 | ||
| Fig. 3 (a) A comparative view of fluorescence intensities of DEAS–BPH (6 μM) upon addition of different metal ions (20 μM) in CH3CN–H2O (4:1 v/v). (b) Metal ion selectivity profile of DEAS–BPH (6 μM): (black bars) change of emission intensity of receptor + 20 μM of Mn+; (red bars) change of emission intensity of receptor + 20 μM of Mn+, followed by 20 μM of Hg2+; λex = 450 nm, λem = 489 nm. | ||
To utilize DEAS–BPH as a selective chemosensor for Hg2+, the effect of other competing metal ions was examined using 20 μM of Hg2+ and 20 μM of the interfering metal ions and is plotted as a bar graph in Fig. 3b. The miscellaneous competitive cations lead to no considerable absorption and fluorescence changes, confirming that the probe retained its binding ability with Hg2+ in the presence of a range of interfering metal ions encountered in environmental and biological settings.
Furthermore, the sensing ability of the receptor with mercury was also investigated at varying pH values from 2.0 to 12.0 by absorption and fluorescence spectral titration (Fig. S10†), and the results revealed that the complex was highly stable at all the pH values. Since the association between DEAS–BPH and Hg2+ remained unaffected within the biologically relevant pH range (5.25–8.93), it is suitable for monitoring of intracellular mercury in living cells without intrusion from pH effects.
Stoichiometry determination and association constant
The binding stoichiometry of the interaction between DEAS–BPH and Hg2+ was quantified by Job's plots based on the absorption titration data (Fig. S11†) which demonstrated the formation of a 2:1 DEAS–BPH:Hg(II) complexation species in the solution (CH3CN–H2O, 4:1, v/v, pH 7.2). The stoichiometry of the complex was further confirmed by ESI-MS analysis, where peak at m/z 942 for the 2:1 complex was observed (Fig. S4b†). The association constant (Ka) as determined from the UV-vis titration was 1.15 × 1010 M−2 and by fluorescence titration method was found to be 1.36 × 1011 M−2 (Fig. S12†), according to the Benesi–Hildebrand equation.18 The detection limits were evaluated to be 3.82 × 10−6 M and 5.57 × 10−7 M through UV-vis (Fig. S13a†) and fluorescence (Fig. S13b†) method,19 respectively and both the results suggested that the receptor was highly sensitive towards mercuric ions.
NMR titration
Further, to elucidate the coordination mode of DEAS–BPH, 1H NMR titration experiment was carried out in DMSO-d6. The spectra of DEAS–BPH before and after the treatment with different equivalent of Hg2+ are presented in Fig. 4, where a prominent change in the chemical shifts of the phenolic proton (Ha) as well as the azomethine proton (Hb) is observed. Upon gradual addition of Hg2+, the –OH proton (Ha) signal underwent a downfield shift from δ 11.27 to a broad signal at δ 11.44 and ultimately disappeared, probably because of the influence of the metal ion. Likewise, the azomethine proton (Hb) experienced a substantial downfield shift from δ 8.69 to δ 8.76 suggesting the direct involvement of azomethine N atom in bonding to Hg2+. A new peak appeared at δ 8.64 adjacent to the azomethine signal which experienced the similar deshielding effect on complexation with Hg2+. Yet again, Hg2+ coordination triggered the aromatic protons to appear at higher magnetic field due to decrease in the electron density upon association to the metal ion, with initiation of new neighbouring peaks. Appearance of the new peaks was complete with the addition of 0.5 equivalent of Hg2+. Thus the NMR titration data substantiated the adduct formation in which the metal cation preferably chelated to the oxygen of the hydroxyl group and the nitrogen of the azomethine group keeping intact the donor part (–NEt2) of the sensor. Hence a downfield shift was observed after binding to Hg2+. | ||
| Fig. 4 1H NMR spectra (400 MHz) measured during the titration of DEAS–BPH with different mole ratio of Hg2+ in DMSO-d6. | ||
Computational studies (DFT)
To obtain an insight into the molecular and electronic structure of the chemosensor DEAS–BPH and its complex with Hg2+, geometry optimizations were performed within the framework of DFT/B3LYP20 method employing the Gaussian 03 program.21 The respective molecular orbital plots of DEAS–BPH and its complex are shown in Fig. S16 and S17 (ESI†) and details of bond distances and bond angles are delineated in Table S1 (ESI†). The optimized structures of the receptor and its complex are provided in Fig. S14† and 5, respectively. | ||
| Fig. 5 Optimized structure of the complex of DEAS–BPH with Hg2+ by DFT/B3LYP/6-31+G(d,p) method. | ||
As evident from Table S1,† the calculated mercury–nitrogen bond distances (105Hg–12N = 2.276 Å and 105Hg–64N = 2.508 Å) are smaller than the sum of the van der Waals radii of Hg and N (2.75 Å).22 Besides, Hg–O bond lengths were calculated to be 2.227 Å (for 105Hg–14O) and 2.382 Å (for 105Hg–66O), respectively, much shorter than the typical covalent bond distance of Hg and O (2.9 Å),23 which clearly supported that the Hg2+ was strongly chelated to the sensor through four coordination sites. The calculated electron distribution in the FMOs revealed that HOMO–LUMO excitation led to displacement of the electron density from the 5-(diethylamino)-2-(iminomethyl) phenol moiety to the diphenylmethanimine units reflecting pronounced ICT character of the fluorophore. Moreover, the HOMO–LUMO energy gap (ΔE = 2.275, Fig. 6) was reduced upon being complexed to Hg2+, compared to the free sensor, thereby accounting for the red-shifted ICT band observed in the absorption spectra.
 | ||
| Fig. 6 Molecular orbital diagram with corresponding energy levels of the complex of DEAS–BPH with Hg2+ in GS and ES calculated at the DFT level using a B3LYP/6-31+G(d,p) basis set. | ||
Time Dependent Density Functional Theory (TDDFT)24 with B3LYP exchange–correlation function was applied to interpret electronic transitions in the compound. The details involving different vertical excitation energies, oscillator strengths and nature of electronic transitions are listed in Table S2 (ESI†).
The theoretically generated UV-vis spectrum of the complex in acetonitrile medium showed a single strong peak at 446 nm which is in accordance with the experimentally obtained value of λmax (448 nm) (Fig. S15†). The absorption band is essentially dominated by the key transitions from HOMO to LUMO, HOMO−1 to LUMO, HOMO to LUMO+1 and HOMO to LUMO+3 (Table S2†).
Reversibility of DEAS–BPH
The reversibility of the binding process between DEAS–BPH and Hg2+ was established when the introduction of excess Na2EDTA (ethylenediaminetetraacetic acid disodium salt) into the solution containing DEAS–BPH (6 μM) and Hg2+ (20 μM) resulted in quenching of the absorbance at 448 nm (Fig. S18†) as well as the emission intensity (Fig. S19†). Because of the strong affinity of EDTA for the Hg2+ ions, demetalation of the receptor–Hg2+ complex occurred causing the absorbance and fluorescence quenching; i.e., Hg2+ ions were not available for chelation with the receptor. Then further addition of Hg2+ ions, immediately revived the absorbance and the fluorescence. Therefore, this study renders the probe as a reversible sensor for the selective recognition of Hg2+ ions in a mixed-solvent medium under physiological conditions.FESEM analysis
In addition, to ascertain whether any morphological alteration of DEAS–BPH has occurred after coordination to Hg2+, FESEM analysis was attempted. Fig. 7a–f show representative FESEM images of the free probe and complexes of DEAS–BPH with 0.5, 1.0 and 2.0 equiv. of Hg(II) respectively. The FESEM images (Fig. 7a and b) demonstrated a rod like microstructure of the probe with an average size of 0.6 ± 0.1 μm in the absence of Hg(II) ions but introduction of Hg2+ initiated shrinkage of the microrods (Fig. 7c). As evident in Fig. 7d, on increasing the concentration of Hg2+, aggregation resulted which led to deformation of the microstructures. This might be accredited to a reorganization of the probe to generate a 2:1 complex with Hg2+.
 | ||
| Fig. 7 FESEM images of DEAS–BPH (a) before addition of Hg2+ and (b) expansion of (a); (c) after addition of 0.5 equiv. Hg2+, (d) after addition of 1.0 equiv. Hg2+, (e) expansion of (d) and (f) after addition of 2.0 equiv. Hg2+. | ||
Cell imaging and cytotoxicity studies
To further evaluate the practical applicability of the probe, fluorescence microscopy images in human hepatocellular liver carcinoma cells (HepG2) were investigated. As shown in Fig. 8, the cells displayed weak green fluorescence when stained with bare DEAS–BPH but after exogenous Hg2+ introduction into the cells, a marked increase in intracellular fluorescence was observed consistent with the results detected in the solution. The followed treatment of the cells with EDTA (50 μM) makes the fluorescence image turn dim distinctly (Fig. 8c), denoting the reduced intracellular Hg2+ level. This experiment indicated that the probe has cell membrane permeability for cell imaging and can be employed as a useful means of detecting intracellular Hg2+ ions in living cells.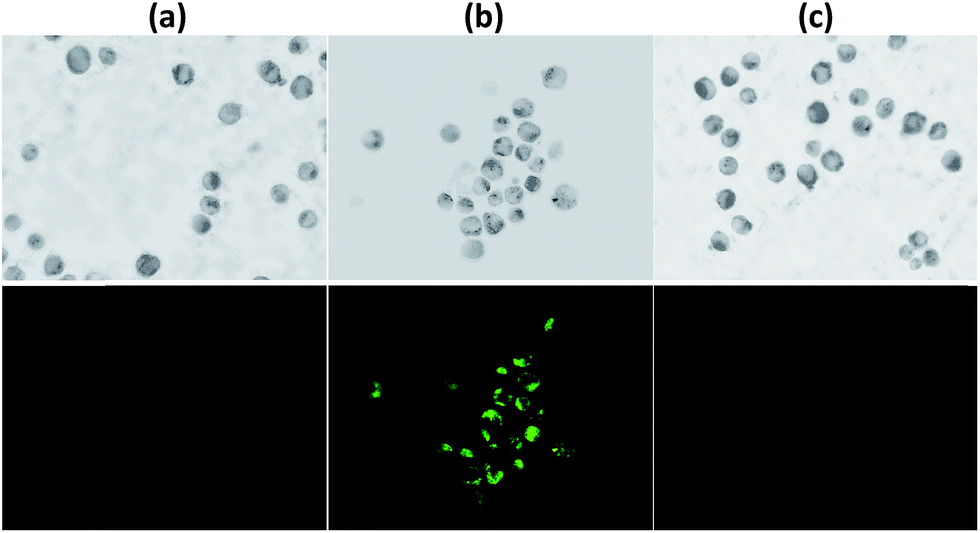 | ||
| Fig. 8 The phase contrast and fluorescence microscopic images of HepG2 cells, (a) cells treated with DEAS–DPH (5 μM), (b) cells treated with DEAS–BPH, further incubated with Hg(NO3)2 (10 μM), (c) cells further treated with EDTA (50 μM). | ||
Moreover, the cytotoxicity of DEAS–BPH toward HepG2 cells was assessed employing the standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay.26 To this end, HepG2 cells were treated with various concentrations of DEAS–BPH (20–80 μM mL−1) for different time intervals 24, 48 and 72 h. As it could be noticed in Fig. 9, that at lower concentration (20 μM mL−1) the chemosensor showed no significant cell death till 72 h, there was a very little rise in cytotoxicity at higher concentrations from 48 h onward. The results clearly suggested that the probe is of low cytotoxicity and can be utilized as an effective mercury monitoring candidate in biological materials (Fig. 10).
 | ||
| Fig. 9 The phase contrast and fluorescence microscopic images of E. coli bacterial cells, (a) cells treated with DEAS–BPH (10 μM), (b) cells treated with DEAS–BPH, further incubated with Hg(NO3)2 (20 μM), (c) cells further treated with EDTA (100 μM). | ||
 | ||
| Fig. 10 % of cell survival of HepG2 cells treated with different concentrations (20–80 μM mL−1) of DEAS–BPH for 24, 48 and 72 h. | ||
Experimental
General
Chemicals were used without further purification as received from commercial suppliers (Sigma Aldrich, Merck) unless otherwise noted. All solvents were dried and distilled prior to use according to the standard protocols. Melting point was determined using a hot-plate melting point apparatus in an open mouth capillary and is uncorrected. The 1H and 13C NMR spectra were recorded on a Bruker 400 MHz and 75 MHz spectrometer respectively, in DMSO-d6 or CDCl3 solutions, using TMS as internal reference (0 ppm). Chemical shifts (δ) are expressed in ppm and all coupling constants (J) are absolute values given in hertz (Hz). The FTIR spectra were recorded with Perkin Elmer Spectrum RXI equipment using KBr pellets. High resolution mass spectrum (HRMS) was acquired using an electron spray ionisation time-of-flight (ESI-TOF) mass spectrometer in positive mode in acetonitrile solvent. UV-vis and fluorescence spectroscopy measurements were carried out using a Shimadzu (model UV1700) spectrophotometer and spectrofluorimeter (model RF 5301) respectively, with quartz cell of path length 10 mm and the nitrate salts of metal were used in the study. Milli-Q water was used throughout the analytical experiments. A field emission scanning electron microscope (FESEM, S4800, Hitachi) equipped with an energy-dispersive X-ray spectrum was applied to determine the morphology.Synthesis of the receptor (DEAS–BPH)
A mixture of 4-N,N-diethylaminosalicylaldehyde 1a (0.300 g, 1.55 mmol) and 1-(diphenylmethylene)hydrazine or benzophenone hydrazone 2 (0.304 g, 1.55 mmol) was dissolved in 15 mL of dry ethanol in presence of 4 drops of acetic acid and the resulting solution was stirred under reflux for 4 h at an ambient temperature. The product precipitated from the reaction mixture and was collected by filtration. It was washed with cold ethanol and dried under vacuum to afford a bright yellow solid (0.472 g, 1.27 mmol, yield = 82%): mp 85–86 °C; 1H-NMR (400 MHz, DMSO-d6): δ 1.06 (t, J = 7.2 Hz, 6H), 3.30–3.36 (m, 4H), 5.94 (d, J = 2.4 Hz, 1H), 6.26 (dd, J1 = 10 Hz, J2 = 2.4 Hz, 1H), 7.22–7.26 (m, 3H), 7.42–7.51 (m, 6H), 7.61–7.63 (m, 2H), 8.69 (s, 1H), 11.27 (s, 1H). 13C-NMR (75 MHz, CDCl3): δ 12.69, 44.62, 97.85, 103.98, 107.30, 128.29, 128.38, 128.72, 128.76, 128.8, 130.21, 133.67, 136.36, 137.81, 151.59, 161.87, 163.44, 165.55. FT-IR (KBr, cm−1): ν 2970, 1633, 1592, 1516, 1413, 1355, 1237, 1131, 791, 692. HRMS (ESI-TOF, m/z): calcd for C24H26N3O [M + H+] 372.2078, found 372.2077.Synthesis of the DEAS–BPH + Hg2+ complex
To a methanolic solution (5 mL) containing the ligand (0.100 g, 0.27 mmol), a methanolic solution of Hg(NO3)2·H2O (0.048 g, 0.14 mmol) was added. The color of the solution changed to bright yellow. The resulting mixture was stirred for 3 h. The solvent was removed under vacuum and the whole mass was washed with ether several times to afford the complex as orange yellow solid (0.140 g, yield = 55%). FT-IR (KBr, cm−1): ν 3430 (broad), 2978, 1600, 1518, 1428, 1339, 1284, 1236, 1138, 1066, 771, 690. HRMS (ESI-TOF, m/z): calculated for C48H48HgN6O2 942.3534, found 942.2808. Elemental analysis calculated (%) for [Hg(DEAS–BPH)2] C48H48HgN6O2 (M.W. 941): C 61.23, H 5.14, N 8.93, O 3.40, found C 61.19, H 5.19, N 8.89.Computational methods
Quantum chemical calculations based on density functional theory (DFT) were carried out using a Gaussian 03 program.21 Geometry optimizations for the ground-state and the first singlet electronic excited state of DEAS–BPH and the DEAS–BPH + Hg2+ complex were computed using the DFT and TD-DFT methods, respectively at the B3LYP level.20,24 The LanL2DZ basis set and core potentials were adopted for mercury while 6-31+G(d,p) basis set for the rest of the atoms.25 The vibrational frequency calculations were executed to ensure that the optimized geometries represent the local minima of potential energy surface with positive eigen-values. The UV spectra were computed from TDDFT calculations in MeCN.Cell culture and imaging
HepG2 (NCCS, Pune, India) cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 mg mL−1 penicillin-G and 50 mg mL−1 streptomycin at 37 °C in a humidified environment containing 5% CO2 inside an incubator (Heal Force, China). Cells were grown up to 80–90% confluence, harvested with 0.025% trypsin (Gibco BRL) and 0.52 mM EDTA (Gibco BRL) in phosphate-buffered saline (PBS, pH 7.4), plated at the desired cell concentration and allowed to re-equilibrate for 24 h prior to treatments. Cells were treated with the probe dissolved in DMSO, while the untreated control cultures received only the vehicle (DMSO, <0.2%). For imaging, cells were rinsed with PBS and incubated with DEAS–BPH (5 μM) for 30 min and then followed by addition of Hg(NO3)2 (10 μM) for another 15 min at 37 °C. The cells were then washed three times with PBS to remove the remaining chemosensor and imaging was carried out using a fluorescence microscope (Leica) under the same experimental condition. Then EDTA (50 μM) was added to the cells, incubated for another 10 min and imaged.E. coli were cultured overnight in Mueller-Hinton broth (MHB) under shaking condition at 37 °C. Cells were harvested by centrifugation at 6000 × g for 5 min with PBS buffer pH 7.4. Cells representing 104 mL−1 concentration was incubated with 10 μM DEAS–BPH for 30 min at 37 °C. Then a solution of 20 μM of Hg(NO3)2 was added onto the cells, and kept at room temperature for 15 min and imaged by a Leica fluorescence microscope. In both cases, the fluorescence signals were monitored at 510 nm after exciting at 480 nm.
In vitro cytotoxicity
MTT assay26 was performed to assess the cytotoxic activity of DEAS–BPH toward HepG2 cells. HepG2 cells were incubated in 96-well sterile plates at a density of 1 × 106 cells per mL with different concentrations (20–80 μM mL−1). The cells were then incubated for 24, 48 and 72 h, respectively. After the designated time intervals, the wells were washed twice with PBS buffer and 100 μL of freshly prepared MTT (0.5 mg mL−1) solution was added into each well. The MTT medium solution was carefully removed after 3 h incubation in the incubator. DMSO (100 μL) was then added into each well and the plate was gently shaken for 10 min at room temperature to dissolve all formazan granules formed by viable cells. The absorbance of MTT at 570 nm was monitored by the microplate reader (Genios Tecan).Conclusions
A simple N,N-diethylaminosalicylaldehyde based reversible, fluorescent chemosensor DEAS–BPH has been developed which exhibited high selectivity and sensitive fluorescence enhancement (∼27 fold) toward mercuric ions on the basis of ICT, over other competing metal ions in mixed aqueous media. The chemosensor allows naked-eye recognition of Hg2+ ions as well as acts as a non-pH-dependent fluorescent probe in the biological pH range. Additionally, the selective sensing of mercuric ion via intramolecular charge transfer (ICT) mechanism is established by showing the non-selective fluorescence output of a similar receptor where DEAS unit is replaced by a salicylaldehyde moiety. Theoretical calculations have been carried out to establish metal–ligand binding mechanism through optimizing their structures. Moreover, the FESEM images provide explicit information on morphological changes of the probe after metal complexation. The cell imaging and MTT assay experiments further demonstrated the cell permeability and negligible cytotoxicity of the chemosensor which make the probe highly utilitarian for the assessment of Hg2+ in biological systems. Thus, the chemosensor meets all the requirements to be an excellent fluorescent probe for wide applications in the field of biolabeling, biosensing, imaging and so on.Acknowledgements
R. R. thanks CSIR, New Delhi, Government of India and S. R acknowledges UGC, New Delhi, Government of India for senior research fellowships. Authors are grateful to Dr K. K. Chattopadhyay, Mr B. Das and Mr S. Kar of Jadavpur University and Mr N. Dutta of Indian Association for the Cultivation of Science for necessary assistance.Notes and references
- (a) A. Renzoni, F. Zino and E. Franchi, Environ. Res., 1998, 77, 68 CrossRef CAS PubMed; (b) O. Malm, Environ. Res., 1998, 77, 73 CrossRef CAS PubMed; (c) H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465 RSC.
- (a) H. H. Harris, I. J. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed; (b) S. Yoon, A. E. Albers, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16030 CrossRef CAS PubMed; (c) E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443 CrossRef CAS PubMed.
- (a) H. H. Harris, I. J. Pickering and G. N. George, Science, 2004, 303, 764 CAS; (b) S. K. Arya and S. Bhansali, Chem. Rev., 2011, 111, 6783 CrossRef CAS PubMed.
- (a) I. Onyido, A. R. Norris and E. Buncel, Chem. Rev., 2004, 104, 5911 CrossRef CAS PubMed; (b) J. Liu, Y.-Q. Sun, P. Wang, J. Zhang and W. Guo, Analyst, 2013, 138, 2654 RSC; (c) S. Pal, J. Hatai, M. Samanta, A. Shaurya and S. Bandyopadhyay, Org. Biomol. Chem., 2014, 12, 1072 RSC.
- (a) N. H. Khdary and A. G. Howard, Analyst, 2011, 136, 3004 RSC; (b) Z. Liu, Z. Zhu, H. Zheng and S. Hu, Anal. Chem., 2013, 84, 10170 CrossRef PubMed; (c) Z. A. Chandio, F. N. Talpur, H. Khan, H. I. Afridi, G. Q. Khaskheli and M. A. Mughal, RSC Adv., 2014, 4, 3326 RSC.
- (a) X. Ai, Y. Wang, X. Hou, L. Yang, C. Zheng and L. Wu, Analyst, 2013, 138, 3494 RSC; (b) M. J. da Silva, A. P. S. Paim, M. F. Pimentel, M. L. Cervera and M. de la Guardia, Anal. Chim. Acta, 2010, 667, 43 CrossRef CAS PubMed.
- (a) S. Ma, Q. Chen, H. Li, P. Wang, S. M. Islam, Q. Gu, X. Yang and M. G. Kanatzidis, J. Mater. Chem. A, 2014, 2, 10280 RSC; (b) H. Zhou, X. Lv, L. Zhang, A. Gong, A. Wu, Z. Liang, G. Peng and H. Lin, J. Mater. Chem. C, 2014, 2, 9625 RSC.
- (a) Z. Zhang, J. Li, X. Song, J. Ma and L. Chen, RSC Adv., 2014, 4, 46444 RSC; (b) T. Yordanova, P. Vasileva, I. Karadjova and D. Nihtianova, Analyst, 2014, 139, 1532 RSC.
- (a) S. J. Lee and M. Moskovits, Nano Lett., 2011, 11, 145 CrossRef CAS PubMed; (b) Y. Du, R. Liu, B. Liu, S. Wang, M.-Y. Han and Z. Zhang, Anal. Chem., 2013, 85, 3160 CrossRef CAS PubMed.
- Z. Gao and X. Ma, Anal. Chim. Acta, 2011, 702, 50 CrossRef CAS PubMed.
- K. Leopold, M. Foulkes and P. Worsfold, Anal. Chim. Acta, 2010, 663, 127 CrossRef CAS PubMed.
- (a) Y. Gao, Z. Shi, Z. Long, P. Wu, C. Zheng and X. Hou, Microchem. J., 2013, 103, 1 CrossRef PubMed; (b) E. P. C. Lai, W. Zhang, X. Trier, A. Georgi, S. Kowalski, S. Kennedy, T. MdMuslim and E. Dabek-Zlotorzynska, Anal. Chim. Acta, 1998, 364, 63 CrossRef CAS.
- (a) E. Ermakova, J. Michalak, M. Meyer, V. Arslanov, A. Tsivadze, R. Guilard and A. Bessmertnykh-Lemeune, Org. Lett., 2013, 15, 662 CrossRef CAS PubMed; (b) H. Son, J. H. Lee, Y.-R. Kim, I. S. Lee, S. Han, X. Liu, J. Jaworski and J. H. Jung, Analyst, 2013, 137, 3914 RSC; (c) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2013, 41, 3210 RSC; (d) B. Hu, L.-L. Hu, M.-L. Chen and J.-H. Wang, Biosens. Bioelectron., 2013, 49, 499 CrossRef CAS PubMed; (e) C. Wang and K. M.-C. Wong, Inorg. Chem., 2013, 52, 13432 CrossRef CAS PubMed; (f) Z. Yan, M.-F. Yuen, L. Hu, P. Sun and C.-S. Lee, RSC Adv., 2014, 4, 48373 RSC.
- (a) D. S. McClure, J. Chem. Phys., 1952, 20, 682 CrossRef CAS PubMed; (b) N. Shao, G.-X. Pang, C.-X. Yan, G.-F. Shi and Y. Cheng, J. Org. Chem., 2011, 76, 7458 CrossRef CAS PubMed; (c) R. Koteeswari, P. Ashokkumar, E. J. P. Malar, V. T. Ramakrishnan and P. Ramamurthy, Chem. Commun., 2011, 47, 7695 RSC; (d) D. Mandal, P. Deb, B. Mondal, A. Thakur, J. Ponniah and S. Ghosh, RSC Adv., 2013, 3, 18614 RSC; (e) K. Zhong, X. Zhou, R. Hou, P. Zhou, S. Hou, Y. Bian, G. Zhang, L. Tang and X. Shang, RSC Adv., 2014, 4, 16612 RSC; (f) K. Kanagaraj, K. Bavanidevi, T. J. Chow and K. Pitchumani, RSC Adv., 2014, 4, 11714 RSC.
- (a) H. N. Kim, S.-W. Nam, K. M. K. Swamy, Y. Jin, X. Chen, Y. Kim, S.-J. Kim, S. Park and J. Yoon, Analyst, 2011, 136, 1339 RSC; (b) P. Xi, L. Huang, G. Xie, F. Chen, Z. Xu, D. Bai and Z. Zeng, Dalton Trans., 2011, 40, 6382 RSC; (c) C. Kaewtong, B. Wanno, Y. Uppa, N. Morakot, B. Pulpoka and T. Tuntulani, Dalton Trans., 2011, 40, 12578 RSC; (d) H.-I. Un, C.-B. Huang, C. Huang, T. Jia, X.-L. Zhao, C.-H. Wang, L. Xu and H.-B. Yang, Org. Chem. Front., 2014, 1, 1083 RSC; (e) M. Tian, L. Liu, Y. Li, R. Hu, T. Liu, H. Liu, S. Wang and Y. Li, Chem. Commun., 2014, 50, 2055 RSC; (f) S. Lee, B. A. Rao and Y.-A. Son, Sens. Actuators, B, 2014, 196, 388 CrossRef CAS PubMed; (g) S. Goswami, S. Maity, A. C. Maity, A. K. Das, B. Pakhira, K. Khanra, N. Bhattacharyya and S. Sarkar, RSC Adv., 2015, 5, 5735 RSC.
- (a) S. Dalapati, S. Jana, M. A. Alam and N. Guchhait, Sens. Actuators, B, 2011, 160, 1106 CrossRef CAS PubMed; (b) X. Xie, X. Chen, B. Li and L. Zhang, Dyes Pigm., 2013, 98, 422 CrossRef CAS PubMed; (c) Y. Li, J. Wu, X. Jin, J. Wang, S. Han, W. Wu, J. Xu, W. Liu, X. Yao and Y. Tang, Dalton Trans., 2014, 43, 1881 RSC; (d) W.-C. Lin, S.-K. Fang, J.-W. Hu, H.-Y. Tsai and K.-Y. Chen, Anal. Chem., 2014, 86, 4648 CrossRef CAS PubMed.
- (a) Y. Chen, C. Zhu, Z. Yang, J. Li, Y. Jiao, W. He, J. Chen and Z. Guo, Chem. Commun., 2013, 48, 5094 RSC; (b) S. Goswami, A. K. Das and S. Maity, Dalton Trans., 2013, 42, 16259 RSC; (c) H. Jiang, J. Jiang, J. Cheng, W. Dou, X. Tang, L. Yang, W. Liu and D. Bai, New J. Chem., 2014, 38, 109 RSC.
- (a) A. J. Benesi and H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef; (b) Y. Shiraishi, S. Sumiya, Y. Kohno and T. Hirai, J. Org. Chem., 2008, 73, 8571 CrossRef CAS PubMed; (c) G. K. Vegesna, J. Janjanam, J. Bi, F.-T. Luo, J. Zhang, C. Olds, A. Tiwari and H. Liu, J. Mater. Chem. B, 2014, 2, 4500 RSC.
- (a) M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414 CrossRef CAS; (b) W. Lin, L. Yuan, Z. Cao, Y. Feng and L. Long, Chem.–Eur. J., 2009, 15, 5096 CrossRef CAS PubMed.
- (a) P. Hohenber and W. Khon, Phys. Rev., 1964, 136, B864 CrossRef; (b) W. Khon and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (d) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 03, revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) A. J. Canty, N. Chaichit, B. M. Gatehouse, E. E. George and G. Hayhurst, Inorg. Chem., 1981, 20, 2414–2422 CrossRef CAS; (b) A. J. Canty, N. Chaichit, B. M. Gatehouse and E. E. George, Inorg. Chem., 1981, 20, 4293–4300 CrossRef CAS.
- (a) S. H. Whitlow, Can. J. Chem., 1974, 52, 198–202 CrossRef CAS; (b) L. G. Kuz'mina, N. G. Bokii and Y. T. Struchkow, Russ. Chem. Rev., 1975, 44, 73–85 CrossRef PubMed.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed; (b) S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS PubMed; (b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS PubMed.
- T. Mossman, J. Immunol. Methods, 1983, 65, 55 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, FT-IR, HRMS spectra, UV-vis and fluorescence titration spectra with different metal ions, association constant and detection limit determination, HOMO and LUMO pictures, Cartesian coordinates. See DOI: 10.1039/c5ra06582h |
| This journal is © The Royal Society of Chemistry 2015 |