
Design and preparation of bio-based dielectric elastomer with polar and plasticized side chains†
Weiwei Leia,
Runguo Wanga,
Dan Yangd,
Guanyi Houa,
Xinxin Zhoua,
He Qiaoa,
Wencai Wanga,
Ming Tiana and
Liqun Zhang*abc
aState Key Laboratory for Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: zhanglq@mail.buct.edu.cn; Fax: +86-10-64456158; Tel: +86-10-64443413
bKey Laboratory of Beijing City for Preparation and Processing of Novel Polymer Materials, Beijing University of Chemical Technology, Beijing 100029, China
cBeijing Engineering Research Center of Advanced Elastomers, Beijing University of Chemical Technology, Beijing 100029, China
dBeijing Key Lab of Special Elastomer Composite Materials, Beijing Institute of Petrochemical Technology, Beijing 102617, China
First published on 21st May 2015
Abstract
A new dielectric elastomer with large actuated strain driven by low electric field was synthesized from di-n-butyl itaconate and isoprene through free radical redox emulsion polymerization. The effect of the copolymerized proportion of poly(di-n-butyl itaconate-co-isoprene) (PDBII) and the dosage of crosslinking agent on the elastic modulus, dielectric properties, and actuated strain of the elastomer were investigated, and a potential dielectric elastomer candidate containing 70 wt% di-n-butyl itaconate was obtained. The permittivity of the PDBII crosslinked by 3.0 phr of dicumyl peroxide was 5.68 at 103 Hz, which was higher than that of commercial acrylic and silicone dielectric elastomers. Without any prestrain, an actuated strain of 20% was obtained at an electric field of 30 kV mm−1. In order to further increase the actuated strain, barium titanate (BaTiO3), a high-dielectric-constant ceramic powder, was utilized to fill the PDBII to form a BaTiO3/PDBII composite. The dielectric constant of the composite increased with increasing content of BaTiO3, and the elastic modulus of the composite was lower than that of the unfilled PDBII, leading to a larger dielectric actuated strain of the composite.
Introduction
Dielectric elastomer is a kind of intelligent multifunctional electroactive polymer. Dielectric elastomers offering performance such as large active strain, fast response, high electromechanical coupling efficiency, light weight, freedom from noise, and low cost1,2 have drawn significant interest in recent years, especially after silicone and acrylic elastomer films with strains in excess of 100% have been reported.3,4 Several applications have already been developed, including artificial muscles,5 sensors,6 micro air vehicles,7 and electrical power generator.8A dielectric elastomer actuator consists of a thin film sandwiched between two compliant electrodes.9 When an electric field is applied, an electrostatic pressure acts on the film, shrinking in the thickness direction and elongating in the planar direction. The actuated strain in the thickness direction is given by the following equation:
 | (1) |
Most studies have focused on the development of high permittivity polymer composites by loading the elastomer matrix with insulating or conductive fillers. The most commonly followed approach utilized high permittivity ceramic fillers in the form of powder, such as barium titanate,12 lead–magnesium niobate,13 and titanium dioxide.14,15 Usually the dielectric improvement needs a high ceramic filler content, which would increase the elastic modulus. Another approach was to prepare percolative composites utilising conductive fillers such as metal nanoparticles,16 polyaniline,17 carbon nanotubes,18,19 and graphite nanoplates.20 The permittivity can dramatically increase as the content of conductive filler approaches the percolation threshold, i.e., less than 20% for a microcapacitor network in a composite. Such percolative composites generally have high dielectric losses near the percolation threshold, mainly because large leakage currents are generated between the connecting conductive fillers. Efforts to modify the fillers to improve the dielectric performance of elastomer composites include the surface modification of BaTiO3 (ref. 21–23) and titanium dioxide (TiO2)24 to get a homogeneous dispersion and the coating of conductive fillers with an insulating layer to avoid the direct contact of conductive fillers with one another. Another way to increase the actuated strain and the dielectric strength of dielectric elastomer is the application of high prestrain,3 which will reduce the thickness of the dielectric elastomer and lead to the alignment of macromolecule chains perpendicular to the applied electric field. However, a rigid frame or supporting structure was needed to maintain the structure of the dielectric film,25 and the stress relaxation and fatigue of the film after prestraining will take place as time goes by. Previous studies mostly focused on the filler and ignored the matrix, but the matrix was also critical for an excellent dielectric elastomer composite to have a low elastic modulus and high dielectric constant. Despite a great many of dielectric elastomer composites have been developed, there was still a severe lack of elastomer with a favorable profile of mechanical and dielectric properties.
The aim of this study was to design and prepare a novel dielectric elastomer based on di-n-alkyl itaconate. Di-n-alkyl itaconic acid ester can be readily polymerized to form a comb-branched structure, which was an excellent candidate for the investigation of ordering in amorphous structures. Nanophase separation of the incompatible main chains and side chains of poly(di-n-alkyl itaconates) has been extensively studied by differential scanning calorimetry (DSC),26 dynamic mechanical analysis (DMA),27,28 dielectric spectroscopy,29 nuclear magnetic resonance (NMR),30,31 and molecular modeling,31 leading to a deeper understanding of the relative roles of intramolecular constraints and intermolecular interactions in the solid-state organization and properties of these polymers. The dielectric elastomers based on di-n-alkyl itaconate are readily polarized, and the alkyl groups provide the nanoheterogeneity and the plasticizing effect.29,31,32 The structure of PDBII is shown in Scheme 1, this structure is similar to that of acrylic elastomer.
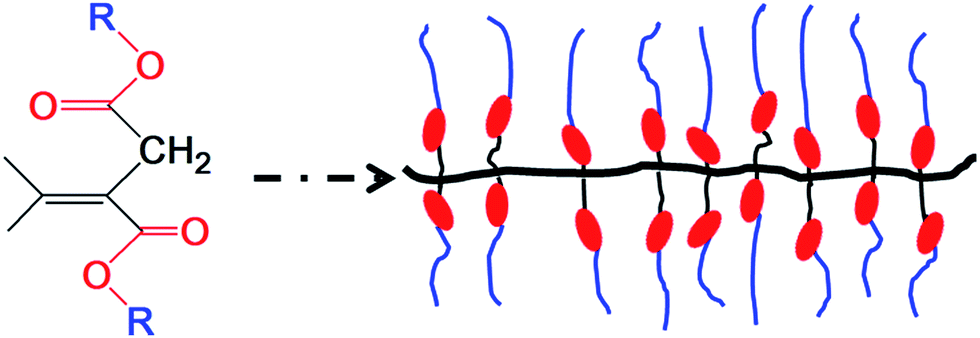 | ||
| Scheme 1 Synthesis of dielectric elastomer with easily polarized ester groups (red circles) and flexible alkyl groups (blue lines) from itaconate. | ||
Itaconic acid, which has been categorized as one of the “top 12” building block molecules in advanced biorefineries,33 is a monomer obtained at low cost from renewable resources by fermentation. Butanol and isoprene were used in systhize PDBII together with itaconic acid. Butanol was used because it has the appropriate side chain length to polymerize a low glass transition temperatures polymer and it is a high-efficiency, renewable biofuel. The isoprene will soften the polymer and provide the crosslinking points. Bio-based isoprene has aroused considerable interest and it is already available in the market. A free radical redox emulsion polymerization method34,35 was used to synthesize PDBII, as we did our previous work.34 The molar ratio of itaconate to isoprene, crosslinking agent dosage, and the content of the high-dielectric ceramic BaTiO3 were studied to modulate the electromechanical properties.
Experimental
Materials
Itaconic acid (purity of 99%) was purchased from Qingdao Langyatai Group Co., Ltd. Butanol (purity of 97%) was purchased from Sigma-Aldrich Company. Isoprene (purity of 95%) was purchased from Alfa Aesar Company and distilled to remove any stabilizer before use. Ferric ethylenediaminetetraacetic acid salt (Fe(II) EDTA), sodium dodecyl benzene sulfonate (SDBS), sodium hydroxymethane sulfonate (SHS), tert-butyl hydroperoxide (TBH), potassium phosphate tribasic, potassium chloride, and hydroxylamine were purchased from Sigma-Aldrich Company and used as received without further purification. The crosslinking agent dicumyl peroxide (DCP) was purchased from Beijing Chemical Reagents Co., Ltd., China. BaTiO3 nanoparticles (average particle size 30 nm) were purchased from Sigma-Aldrich Company. All the other agents were commercial products without further purification.Methods
| Ingredients (concentration) | Amount (g) |
|---|---|
| a Di-n-butyl itaconate and isoprene were copolymerized in mass ratios of 70/30 (PDBII-70), 50/50 (PDBII-50), and 30/70 (PDBII-30). Polyisoprene was also synthesized for comparison. | |
| Di-n-butyl itaconate | Variablea |
| Isoprene | Variablea |
| Deionized water | 30% solid content |
| SDBS solution (10%) | 5% weight of monomers |
| Potassium phosphate tribasic solution (10%) | 2 |
| Potassium chloride (10%) | 5 |
| SHS solution (10%) | 2 |
| Fe(II)EDTA solution (1%) | 4 |
| TBH solution (10%) | 0.5 |
| Hydroxylamine solution (50%) | 0.4 |
Measurements
| Sp = (A − A0)/A0 × 100% | (2) |
Based on the law of volume constancy,
| (1 + Sp)(1 + Sz) = 1 | (3) |
Rearrangement of eqn (3) gives an expression for the planar strain:
 | (4) |
According to eqn (1),
| Sp = εrε0U/(dY − εrε0U) | (5) |
Every experimental data point of mechanical properties, dielectric properties, and electromechanical strain in this work was the average of the results obtained from at least five samples under the same conditions.
Results and discussion
Synthesis and structure of di-n-butyl itaconate
Itaconic acid was capped with butanol, a renewable monomer, to restrain the chain the chain transfer effect of carboxyl groups. In Fig. S1,† the resonances at 5.69 and 6.32 ppm are both assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 protons of itaconate, indicative of double bonds for subsequent reaction. Absorptions at 4.17, 4.10, 1.63, 1.61, 1.40 and 1.38 ppm are due to the d, e, f, g, h, i protons of the methylenes groups of the capped product, respectively. The absorptions at 0.95 and 0.93 ppm belong to the –CH3 protons of butyl units. In conclusion, di-n-butyl itaconate was successfully produced.
CH2 protons of itaconate, indicative of double bonds for subsequent reaction. Absorptions at 4.17, 4.10, 1.63, 1.61, 1.40 and 1.38 ppm are due to the d, e, f, g, h, i protons of the methylenes groups of the capped product, respectively. The absorptions at 0.95 and 0.93 ppm belong to the –CH3 protons of butyl units. In conclusion, di-n-butyl itaconate was successfully produced.
Molecular weight and structural characteristics of PDBII
The PDBIIs with different di-n-butyl itaconate content were synthesized, and Fig. 1(e) shows photographs of the dry PDBIIs. As the isoprene content increased, the polymer became less consolidated and soft. The GPC results listed in Fig. S2† showed three traces with broad single peaks suggesting only one component exists. The number-average molecular weight of PDBII-70, PDBII-50, PDBII-30 were 19.3 × 104, 18.1 × 104, and 12.3 × 104 respectively, ensuring a sufficiently high elasticity for PDBII to be used as elastomers after being crosslinked.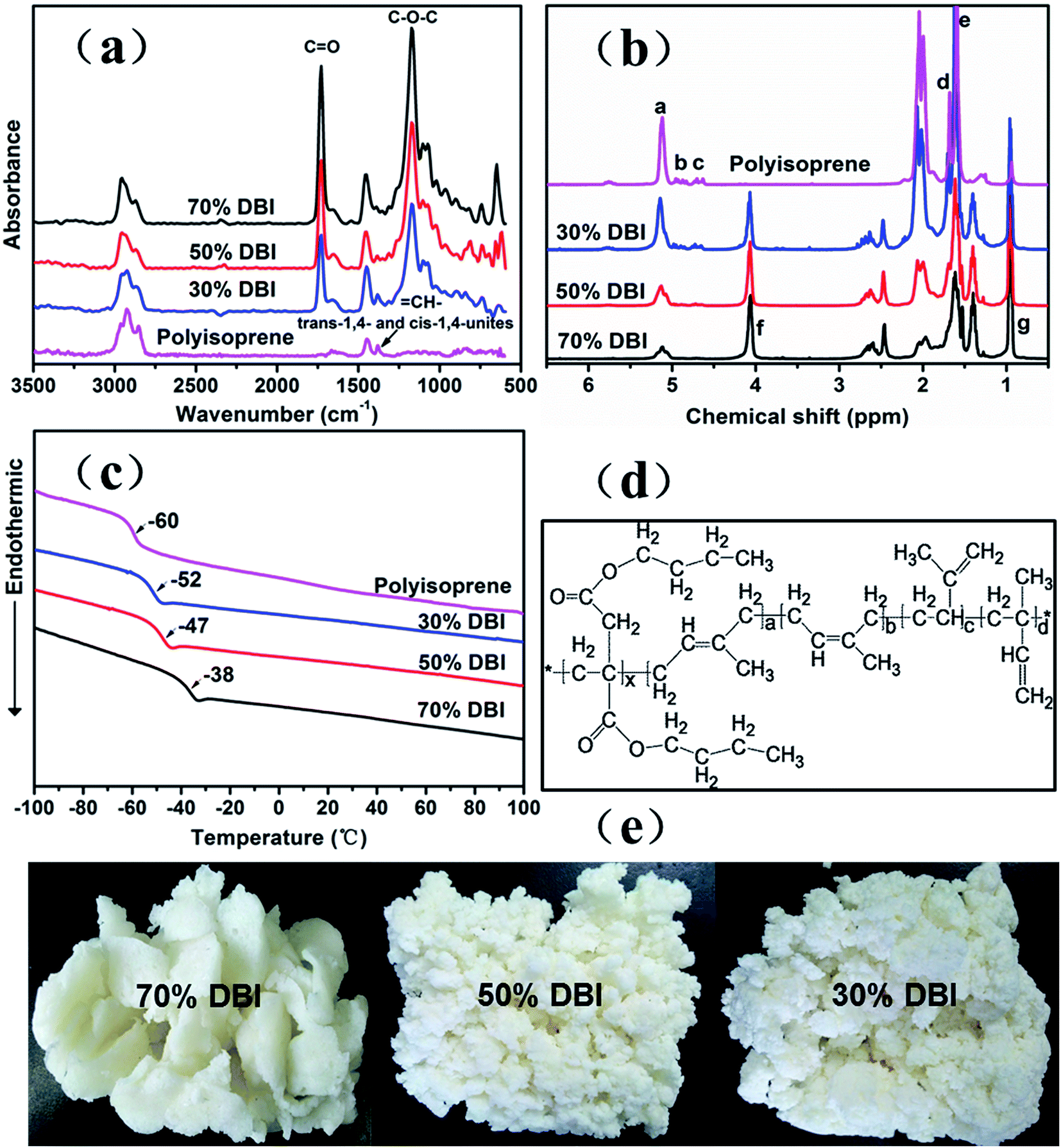 | ||
| Fig. 1 Samples of PDBII with different itaconate to isoprene feed ratios: (a) FTIR spectra, (b) 1H-NMR spectra, (c) DSC thermograms, (d) chemical structure, and (e) sample pictures (DBI: di-n-butyl itaconate, i.e., 30% DBI means 30 wt % DBI in the feed). | ||
As shown in Fig. 1(a), although the most of the absorbance peaks of polyisoprene are overlapping, the peak at 1380 cm−1 representing the trans-1,4- and the cis-1,4-units of isoprene in the main chain can still be distinguished, and the intensity decreases as the di-n-butyl itaconate (DBI) content increases from 30% to 70%. The peak at 1730 cm−1 representing the carbonyl moieties (CO) and the peak at 1170 cm−1 representing the asymmetric vibration of C–O–C indicate that DBI has been successfully copolymerized. Because most of the CH3 and CH2 absorption peaks are overlapping, the detailed structure of PDBII, in particular the copolymerization ratio, should be confirmed by 1H-NMR.
In Fig. 1(b), the polyisoprene proton signals at 5.12 ppm (CH–, peak a), 4.88 ppm (CH2, peak b), and 4.67 ppm (CH2, peak c) represent the 1,4-units, 1,2-units, and 3,4-units, respectively. The peak areas indicate that the polyisoprene prepared by redox emulsion polymerization consists of 93% 1,4-units, 3.3% 1,2-units, and 3.7% 3,4-units. The area under peak e (1.59 ppm, –CH3) is larger than that under peak d (1.68 ppm, –CH3) because there are more trans-1,4-units than cis-1,4-units. The sharp peak g (0.92 ppm, –CH3) corresponds to the –CH3 in the DBI unit. From the areas under peaks g and a, we can deduce that PDBII-70, PDBII-50, and PDBII-30 consist of 72.0%, 57.7%, and 39.8% DBI, respectively. The chemical structures of PDBIIs are summarized in Fig. 1(d).
Thermal properties such as crystallization and glass transition are pivotal for dielectric elastomers. As shown in Fig. 1(c), the PDBII and polyisoprene synthesized by redox emulsion polymerization are amorphous in the testing range of temperature, and the glass transition temperatures decreases with increasing isoprene content.
Effect of molar ratio of di-n-butyl to isoprene
The curing characters of PDBIIs with different DBI content in the feed were listed in Fig. S3 and Table S2.† The frequency dependence of dielectric constant and loss tangent of crosslinked PDBII in the frequency range of 10 to 106 Hz is shown in Fig. 2. The dielectric constant increases with increasing DBI content over the whole frequency range. As there are many polarizable ester groups on the side chains, DCP-crosslinked PDBII with 70% DBI exhibits a high dielectric constant of 5.6 at 103 Hz, which is higher than those of two commonly used dielectric elastomers: silicone elastomer (Nusil CF19-2186, 2.8)3 and acrylic elastomer (3 M VHB, about 4.8)3. Generally, the more abundant the ester groups on the polymer side chain, the higher the dielectric constant of the polymer. The alkyl groups on the side chain always provide some plasticizing effect, but the amount of DBI on the polymer chain have to be optimized to guarantee a high elasticity. Thus, crosslinked PDBII with a DBI content of 70% will be selected in the following research. | ||
| Fig. 2 Frequency dependence of (a) dielectric constant and (b) loss tangent of crosslinked PDBII. (70% DBI stands for 70% DBI in the feed). | ||
The actuated strain of a dielectric elastomer under a given voltage is an important property. The actuated plane strain of PDBII as a function of applied electric field is shown in Fig. 3. It can be seen that the actuated strain increases as the electric field increases. PDBII with 70% DBI displays a higher actuated strain than PDBII with 50% or 30% because the ester groups have strong polarity, which enhances the dielectric constant, and PDBII with 70% DBI has fewer double bonds and hence a lower crosslink density than PDBII with a lower DBI content.
 | ||
| Fig. 3 Actuated strain of crosslinked PDBII at different DBI contents. | ||
Effect of crosslink density
The tensile strength, elastic modulus, electric breakdown strength, actuated strain, and response speed after stimulation are closely related to the crosslink density. Meanwhile, a high actuated strain at a low electric field is actively pursued. PDBII with a DBI content of 70%, crosslinked with different amounts of DCP, were further investigated. The Fig. S4† shows that the PDBII is effectively crosslinked more than 1.5 phr of DCP in the PDBII matrix, and the optimum cure time were summarized in Table S3.† And the torque at optimum cure time increases as the DCP dosage increases.Fig. 4(a) shows the frequency dependence of dielectric constant of PDBII at ambient temperature. With increasing DCP content, the dielectric constant first increases, probably because the free volume of the material decreases with increasing degree of crosslinking.36 However, the dielectric constant decreases with further increases of DCP dosage because a high crosslink density limits the polarization of the polymer chains. Meanwhile, the dielectric constant decreases significantly with increasing frequency at frequencies above 104 Hz because the polarization of the polymer chains do not have enough relaxation time to catch up with the frequency change of an applied electric field.37 Fig. 4(b) shows that the crosslink density has no significant effect on the loss tangent.
 | ||
| Fig. 4 Frequency dependence of (a) dielectric constant and (b) loss tangent of PDBII (with 70% DBI) crosslinked by different amounts of DCP. | ||
Fig. 5 shows that the elongation at break decreases from 623% to 51% and the elastic modulus increases almost linearly from 0.092 MPa to 0.55 MPa with increasing DCP dosage. The modulus of crosslinked PDBII is of the same order of magnitude as two commercial dielectric elastomers: VHB 4910 (ref. 38) (about 0.1 MPa) and DC 3481 (ref. 7) (Dow Corning SILASTIC 3481, about 0.35 MPa). With about the same elastic modulus, the PDBII dielectric elastomers can load about the same stress as the two commercial dielectric elastomers. However, further increases in elastic modulus will lead to a low driving factor for dielectric elastomer, which is defined as the ratio of dielectric constant to elastic modulus.
 | ||
| Fig. 5 Stress–strain curves of PDBIIs (70% DBI) crosslinked by different contents of DCP and (inset) plots of elastic modulus versus DCP dosage and ratio of dielectric constant to elastic modulus versus DCP dosage. | ||
The actuated strains of PDBIIs with different contents of DCP are shown in Fig. 6. With decreasing DCP content, the actuated strain at a given electric field dramatically increases, mainly because a decrease in DCP content reduces the elastic modulus and thus increases the ratio of dielectric constant to elastic modulus. The highest actuated strain at 20 kV mm−1, 30 kV mm−1, and 42 kV mm−1 are 14.7%, 20.0%, and 25.5%, respectively, without any prestrain. This actuated strain is relatively high compared with that of reported polymer dielectric materials under the condition of no prestrain (see ESI†). However, the electric breakdown strength increases with increasing content of DCP, as shown in Fig. 6(a), probably because of the decrease in sample defects with increasing DCP content.
 | ||
| Fig. 6 (a) Actuated strain of PDBII (70%DBI) crosslinked by different contents of DCP and (b) comparison between experimental and theoretical values of actuated strain. | ||
Fig. 6(b) compares the theoretical and experimental actuated strains of PDBII dielectric elastomer. The theoretical planar actuated strain Sp was calculated by eqn (5). The theoretical values are higher than the experimental values under the same applied electric field. During the process of actuation, a small part of the electric energy is converted into thermal energy by mechanical loss and dielectric loss. The discrepancy between the experimental and theoretical values increases with increasing electric field because the mechanical loss increases as the actuated strain increasing and thus more electric energy is transformed into thermal energy, in agreement with previous studies.39,40
BaTiO3/PDBII composites
Fig. 7 show the curing characteristics and stress–strain curves of BaTiO3/PDBII composites. Usually, a polymer composite filled with inorganic nanoparticles has a higher elastic modulus and a lower elongation at break than the polymer matrix, and the elastic modulus increases and the elongation at break decreases with increasing filler content. The inorganic nanoparticles act as physical crosslinks in the polymer matrix to cause a reinforcing effect and hinder the mobility of polymer chains. However, as shown in Fig. 7(b), the PDBIIs filled with 10 to 70 phr of BaTiO3 have lower elastic modulus and higher elongation at break than the pure PDBII. Elastic modulus and curing character were summarized in Table S4.† Similar results were first reported by Carpi14 for silicone dielectric elastomer filled with titanium dioxide. Furthermore, as the BaTiO3 dosage increases to 90 phr, the elongation at break increases sharply to higher than 400% and the elastic modulus comes close to that at a BaTiO3 dosage of 10 phr.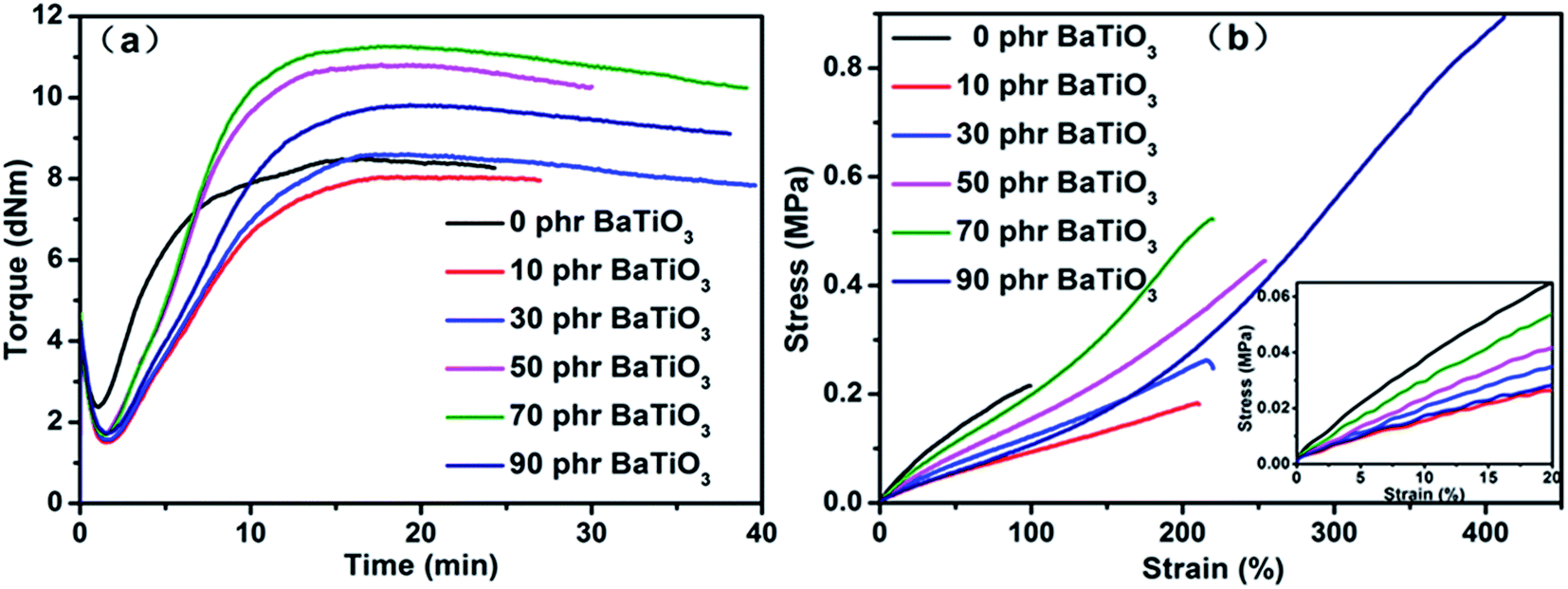 | ||
| Fig. 7 (a) Curing curves and (b) stress–strain curves of BaTiO3-filled PDBIIs. | ||
This unusual phenomena are mainly attributed to the decrease in crosslink density. The crosslink density can be illustrated by the torque increment (ΔS) between the maximum torque (Smax) and minimum torque (Smin) in the curing curve. The curing curves of PDBII elastomers with different dosage of BaTiO3 are shown in Fig. 7(a), and the data of Smax, Smin, ΔS, optimum cure time and elastic modulus are summarized in Table S4.† As reported previously, the elastic modulus of BaTiO3/PDBII is controlled by two factors: the softening effect as a result of reduced crosslinking and the hardening effect of the reinforcement of the filler network.41 Two mechanisms for the decrease in crosslink density in TiO2-filled dielectric elastomers were proposed: polymer chain cleavage at the TiO2 particles42 and the combination of the radicals produced by the TiO2 particles with the radicals produced by DCP.41 Although the pure PDBII presents the highest elastic modulus (see Fig. 7(b)) as a result of high crosslink density, the torque of the filled PDBIIs (see Fig. 7(a)) and the elastic modulus (see Fig. 7(b)) increase as the BaTiO3 dosage increases from 10 phr to 70 phr because of the filler hardening effect. The lower elastic modulus and higher elongation at break at 90 phr of BaTiO3 than at lower dosages were probably due to secondary structures such as nanoparticle aggregates, which further hindered the crosslinking.
The Scanning electron microscope and transmission electron microscope micrographs of BaTiO3/PDBII composites are shown in Fig. 8(a) and (b). The nanoparticles in the diameter range of 100–200 nm are uniformly dispersed in the PDBII matrix. However, the nanoparticle tended to aggregate even at only 10 phr. If the aggregates are sufficiently large and/or numerous, the particles will come into direct contact with one another to form agglomerates (circled in red).43 An obvious filler network appears in Fig. 8(b6) for the composite with 90 wt% BaTiO3, in which the crosslink reaction was most hindered.
 | ||
| Fig. 8 (a) Scanning electron microscope and (b) transmission electron microscope micrographs of PDBII elastomers filled with different contents of BaTiO3. | ||
A simplified model of BaTiO3/PDBII composites is shown in Fig. 9. The high surface energy of BaTiO3 nanoparticles results in strong physical adsorption and/or chemisorption, which make it easy for the nanoparticles to form aggregates. As a result, single particles are rare even at low contents of BaTiO3, as shown in Fig. 8(a2), (b2), and 9(a). As the BaTiO3 content increases, secondary filler structures or filler network which may also be described as agglomerates are formed. However, filler networks on a macro scale are not formed until the BaTiO3 exceeds the percolation threshold of 90 phr. Some polymer chains are trapped in the filler networks without being crosslinked. At moderate and high strains, uncrosslinked rubber is released from filler network as the agglomerates are destroyed, and acts as the polymer matrix. As a result, the elastic modulus of PDBII filled with 90 phr of BaTiO3 decreases and the elongation at break increases sharply, as shown in Fig. 7(b).
 | ||
| Fig. 9 Schematic diagram of dispersion of different weight contents of BaTiO3 in PDBII elastomer: (a) 10%, (b) 50%, and (c) 90%. | ||
Fig. 10(a) shows the frequency dependence of the dielectric constant of BaTiO3/PDBII elastomer composites with different contents of BaTiO3. The dielectric constant increases with increasing content of BaTiO3 particles at all frequencies because of the enhancement of electron polarization and interface polarization at the interface between the PDBII matrix and BaTiO3 particles.44 However, the electron polarization and interface polarization will increase not only the dielectric constant, but also the dielectric loss of the composites. Fig. 10(b) shows the frequency dependence of the dielectric loss of the PDBII elastomer composites at room temperature. The dielectric loss of pure PDBII is smaller than those of all BaTiO3/PDBII composites. As explained above, the semiconductor nature of BaTiO3 is beneficial to the movement of electrical charge, leading to an increase in dielectric loss of BaTiO3/PDBII composites. Besides, the dielectric loss of the composites increases rapidly with frequency at frequencies higher than 104 Hz.
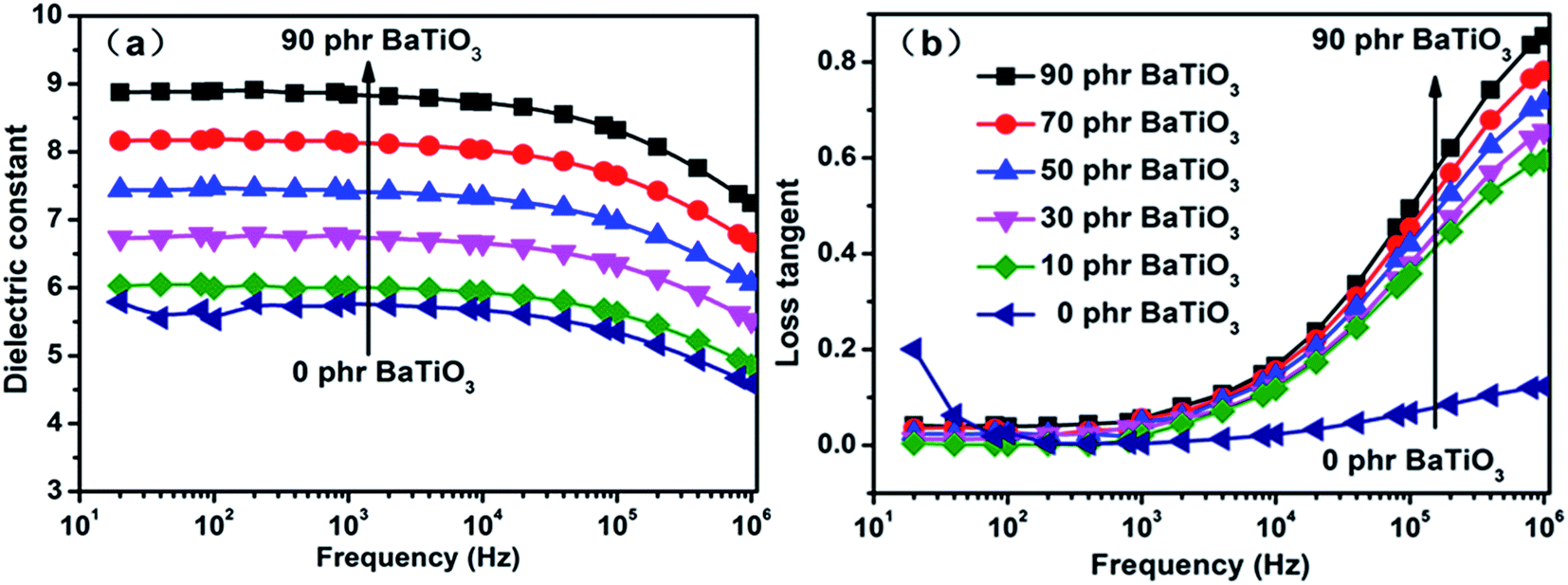 | ||
| Fig. 10 Frequency dependence of (a) dielectric constant and (b) loss tangent of BaTiO3-filled PDBIIs. | ||
The theoretical and experimental actuated strains of PDBII elastomer composites with different contents of BaTiO3 are plotted against the applied electric field Fig. 11. There is a local maximum in actuated strain at an applied electric field of 15 kV min−1 for the BaTiO3/PDBII composite with 10 wt% of BaTiO3. This local maximum in actuated strain of 8.0% was obtained without any prestrain and is 321% higher than the actuated strain of pure PDBII at the same applied electric field. An electric field of 29.2 kV mm−1 was needed to obtain almost the same actuated strain with pure PDBII elastomer; that is, a reduction of 48.6% in electric field was obtained with the composite. As the BaTiO3 content increases from 10 wt% to 70 wt%, the actuated strain decreases because of a decrease in electromechanical sensitivity (β = εr/Y). And the experimental data are in good agreement with theoretical predictions. However, the actuated strain increases sharply as the BaTiO3 content increases from 70 wt% to 90 wt%, mainly because the decrease in elastic modulus discussed above. The discrepancy between experimental data and theoretical prediction increases sharply because filler networks increase the conversion of electric energy to thermal energy through mechanical loss and dielectric loss.
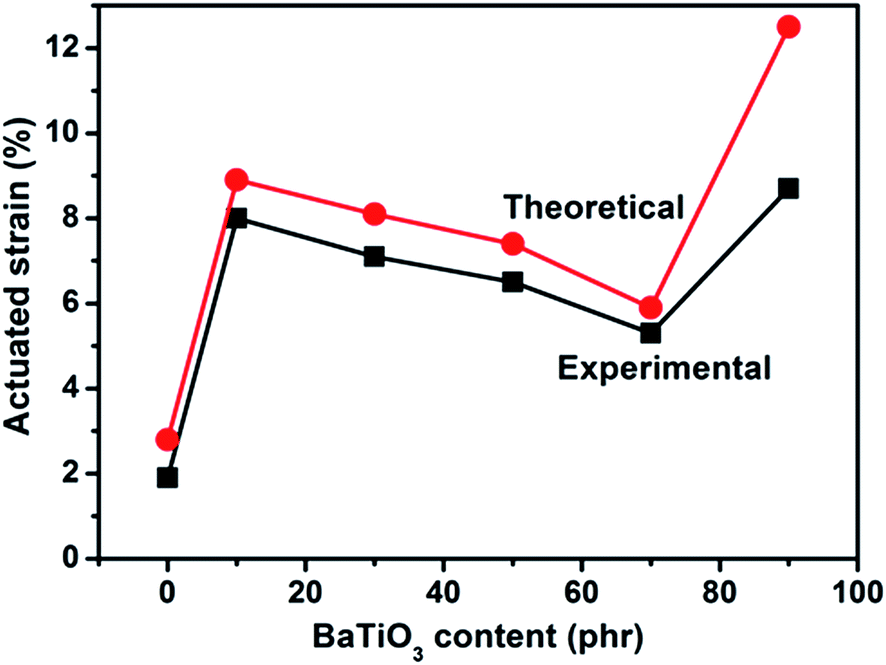 | ||
| Fig. 11 Comparison of theoretical (red dots) and experimental (black squares) actuated strains of BaTiO3/PDBII composites at 15 kV mm−1. | ||
Conclusion
The novel poly(di-n-butyl itaconate-co-isoprene) (PDBII) dielectric elastomer have been synthesized from itaconic acid, butanol, and isoprene by free radical redox emulsion polymerization. After being crosslinked by 3.0 phr of dicumyl peroxide (DCP), PDBII with a di-n-butyl itaconate content of 70%, a number-average molecular weight of 310![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and a glass transition temperature of −38 °C showed a permittivity of 5.68 at 103 Hz. The effect of DCP content on the mechanical, dielectric, and actuation properties was investigated. An actuated strain of 20% at an electric field of 30 kV mm−1 and an actuated strain of 25% at an electric field of 43 kV mm−1 were obtained with PDBIIs crosslinked with 3.0 and 5.0 phr of DCP, respectively, without any prestrain. PDBII (mass ratio of di-n-butyl itaconate to isoprene of 70/30) filled with 10.0 phr of barium titanate and crosslinked by 5.0 phr of dicumyl peroxide had an actuated strain of 8.0% at 15 kV mm−1, 321% higher than the actuated strain of pure PDBII. This research may help establish a new and valuable route for dielectric elastomers.
000, and a glass transition temperature of −38 °C showed a permittivity of 5.68 at 103 Hz. The effect of DCP content on the mechanical, dielectric, and actuation properties was investigated. An actuated strain of 20% at an electric field of 30 kV mm−1 and an actuated strain of 25% at an electric field of 43 kV mm−1 were obtained with PDBIIs crosslinked with 3.0 and 5.0 phr of DCP, respectively, without any prestrain. PDBII (mass ratio of di-n-butyl itaconate to isoprene of 70/30) filled with 10.0 phr of barium titanate and crosslinked by 5.0 phr of dicumyl peroxide had an actuated strain of 8.0% at 15 kV mm−1, 321% higher than the actuated strain of pure PDBII. This research may help establish a new and valuable route for dielectric elastomers.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant nos 50933001, 51173007 and 51221002).References
- P. H. Vargantwar, A. E. Oezcam, T. K. Ghosh and R. J. Spontak, Adv. Funct. Mater., 2012, 22, 100–2113 CrossRef PubMed.
- Y. Jang and T. Hirai, Soft Matter, 2011, 7, 10818–10823 RSC.
- R. Pelrine, R. Kornbluh, Q. Pei and J. Joseph, Science, 2000, 287, 836–839 CrossRef CAS.
- R. Kornbluh, R. Pelrine, Q. Pei, S. Oh and J. Joseph, Proc. SPIE-Int. Soc. Opt. Eng., 2000, 3987, 51–64 CAS.
- P. Brochu and Q. Pei, Macromol. Rapid Commun., 2010, 31, 10–36 CrossRef CAS PubMed.
- F. Carpi, S. Bauer and D. De Rossi, Science, 2010, 330, 1759–1761 CrossRef CAS PubMed.
- X. Q. Zhang, C. Lowe, M. Wissler, B. Jahne and G. Kovacs, Adv. Eng. Mater., 2005, 7, 361–367 CrossRef CAS PubMed.
- S. Chiba, M. Waki, R. Kornbluh and R. Pelrine, Smart Mater. Struct., 2011, 20, 124006 CrossRef.
- M. Molberg, D. Crespy, P. Rupper, F. Nueesch, J. A. E. Manson, C. Loewe and D. M. Opris, Adv. Funct. Mater., 2010, 20, 3280–3291 CrossRef CAS PubMed.
- F. Carpi, G. Gallone, F. Galantini and D. De Rossi, Adv. Funct. Mater., 2008, 18, 235–241 CrossRef CAS PubMed.
- G. Gallone, F. Galantini and F. Carpi, Polym. Int., 2010, 59, 400–406 CrossRef CAS PubMed.
- K. H. Lee, J. Kao, S. S. Parizi, G. Caruntu and T. Xu, Nanoscale, 2014, 6, 3526–3531 RSC.
- G. Gallone, F. Carpi, D. De Rossi, G. Levita and A. Marchetti, Mater. Sci. Eng., C, 2007, 27, 110–116 CrossRef CAS PubMed.
- F. Carpi and D. De Rossi, IEEE Trans. Dielectr. Electr. Insul., 2005, 12, 835–843 CrossRef CAS.
- D. Yang, L. Zhang, N. Ning, D. Li, Z. Wang, T. Nishi, K. Ito and M. Tian, RSC Adv., 2013, 3, 21896–21904 RSC.
- Y. Shen, Y. Lin, M. Li and C.-W. Nan, Adv. Mater., 2007, 19, 1418–1422 CrossRef CAS PubMed.
- J. K. Yuan, Z. M. Dang, S. H. Yao, J. W. Zha, T. Zhou, S. T. Li and J. Bai, J. Mater. Chem., 2010, 20, 2441–2447 RSC.
- Z. M. Dang, L. Wang, Y. Yin, Q. Zhang and Q. Q. Lei, Adv. Mater., 2007, 19, 852–857 CrossRef CAS PubMed.
- K. Goswami, A. E. Daugaard and A. L. Skov, RSC Adv., 2015, 5, 12792–12799 RSC.
- M. Tian, Z. Wei, X. Zan, L. Zhang, J. Zhang, Q. Ma, N. Ning and T. Nishi, Compos. Sci. Technol., 2014, 99, 37–44 CrossRef CAS PubMed.
- P. Kim, S. C. Jones, P. J. Hotchkiss, J. N. Haddock, B. Kippelen, S. R. Marder and J. W. Perry, Adv. Mater., 2007, 19, 1001–1005 CrossRef CAS PubMed.
- D. Yang, M. Tian, D. Li, W. Wang, F. Ge and L. Zhang, J. Mater. Chem. A, 2013, 1, 12276–12284 CAS.
- K. Hayashida, Y. Matsuoka and Y. Takatani, RSC Adv., 2014, 4, 33530–33536 RSC.
- H. Stoyanov, M. Kollosche, S. Risse, D. N. McCarthy and G. Kofod, Soft Matter, 2011, 7, 194–202 RSC.
- S. M. Ha, W. Yuan, Q. B. Pei, R. Pelrine and S. Stanford, Adv. Mater., 2006, 18, 887–891 CrossRef CAS PubMed.
- J. Cowie, I. McEwen and M. Y. Pedram, Macromolecules, 1983, 16, 1151–1155 CrossRef CAS.
- J. Cowie, S. Henshall and I. McEwen, Polymer, 1977, 18, 612–616 CrossRef CAS.
- J. Cowie, Z. Haq, I. McEwen and J. Velickovic, Polymer, 1981, 22, 327–332 CrossRef CAS.
- V. Arrighi, I. McEwen and P. Holmes, Macromolecules, 2004, 37, 6210–6218 CrossRef CAS.
- A. C. Genix and F. Lauprêtre, Macromolecules, 2006, 39, 7313–7323 CrossRef CAS.
- A. C. Genix and F. Lauprêtre, J. Non-Cryst. Solids, 2006, 352, 5035–5041 CrossRef CAS PubMed.
- V. Arrighi, A. Triolo, I. McEwen, P. Holmes, R. Triolo and H. Amenitsch, Macromolecules, 2000, 33, 4989–4991 CrossRef CAS.
- W. J. Orts, K. M. Holtman and J. N. Seiber, J. Agric. Food Chem., 2008, 56, 3892–3899 CrossRef CAS PubMed.
- R. Wang, J. Ma, X. Zhou, Z. Wang, H. Kang, L. Zhang, K. Hua and J. Kulig, Macromolecules, 2012, 45, 4989–4991 CrossRef.
- Z. Liu, Y. Han, C. Zhou, M. Zhang, W. Li, H. Zhang, F. Liu and W. J. Liu, Ind. Eng. Chem. Res., 2010, 49, 7152–7158 CrossRef CAS.
- G. Hougham, G. Tesoro and A. Viehbeck, Macromolecules, 1996, 29, 3453–3456 CrossRef CAS.
- Z. M. Dang, H. P. Xu and H. Y. Wang, Appl. Phys. Lett., 2007, 90, 2901–12903 Search PubMed.
- D. Yang, M. Tian, H. Kang, Y. Dong, H. Liu, Y. Yu and L. Zhang, Mater. Lett., 2012, 76, 229–232 CrossRef CAS PubMed.
- H. Liu, L. Zhang, D. Yang, Y. Yu, L. Yao and M. Tian, Soft Mater., 2013, 11, 363–370 CrossRef CAS PubMed.
- D. De Tommasi, G. Puglisi and G. Zurlo, Appl. Phys. Lett., 2011, 98, 125507, DOI:10.1063/1.356888.
- D. Yang, M. Tian, Y. Dong, H. Kang, D. Gong and L. Zhang, J. Appl. Phys., 2013, 114, 154104 CrossRef PubMed.
- Y. M. Xu and C. H. Langford, Langmuir, 2001, 17, 897–902 CrossRef CAS.
- Z. Wang, J. Liu, S. Wu, W. Wang and L. Zhang, Phys. Chem. Chem. Phys., 2010, 12, 3014–3030 RSC.
- Z. M. Dang, Y. J. Xia, J. W. Zha, J. K. Yuan and J. Bai, Mater. Lett., 2011, 65, 3430–3432 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06576c |
| This journal is © The Royal Society of Chemistry 2015 |