
Sulfonated polyethylene glycol (PEG-OSO3H) as a polymer supported biodegradable and recyclable catalyst in green organic synthesis: recent advances
Abstract
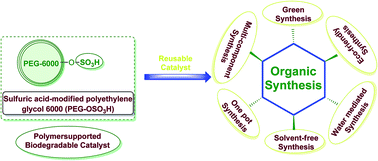
The objective of this review is to summarize some of the recent advances in a sulfuric acid-modified polyethylene glycol 6000 (PEG-OSO3H) as a stable, recyclable and bio-degradable polymeric catalyst in organic synthesis. PEG-OSO3H is a highly efficient and eco-friendly catalyst, which catalyzed various organic reactions and could be recovered and reused several times without significant loss of its activity. In this review, preparation and application of PEG-OSO3H in organic synthesis are investigated.
 Please wait while we load your content...
Please wait while we load your content...

