DOI:
10.1039/C5RA06500C
(Paper)
RSC Adv., 2015,
5, 50611-50616
Tunable color and energy transfer of Tm3+ and Ho3+ co-doped NaGdF4 nanoparticles
Received
11th April 2015
, Accepted 28th May 2015
First published on 28th May 2015
Abstract
NaGdF4:Tm3+, Ho3+ nanoparticles with luminescence properties were synthesized by a hydrothermal method. The morphology, structure and properties of the obtained samples were investigated in detail. The results indicate that the samples are pure hexagonal NaGdF4. The luminescence properties reveal that the samples can emit blue and green light under 359 nm UV light excitation. It is found that in the Tm3+, Ho3+ co-doped NaGdF4 system, the efficient energy transfer from Tm3+ to Ho3+ is observed from the photoluminescence spectra and the fluorescence decay curves. Furthermore, the hues can be adjusted from blue through to light blue and ultimately to bluish green. It is clear that these β-NaGdF4 nanoparticles could have potential applications in fields such as solid-state lighting and color displays.
1 Introduction
In recent years, an increasing number of researchers have a keen interest in studying and developing luminescent nanomaterials such as lanthanide-doped oxides,1–3 phosphates,4–7 and fluorides,8–10 by reason of their distinct emission properties deriving from the 4f electron configuration.11 Among all kinds of host materials for lanthanide-doped nanocrystals, rare-earth fluoride (such as NaGdF4) nanocrystals have garnered much more interest on account of their outstanding properties, for example, low phonon energy, high refractive index and chemical stability, and relatively low crystallization temperature.12 Therefore, many different rare earth-doped NaGdF4 nanoparticles have been obtained, such as NaGdF4:Eu3+,13 NaGdF4:Ce3+/Ln3+ (Ln = Tb, Eu, Dy),14 NaGdF4:Tb3+, Sm3+,15 NaGdF4:Yb3+, Er3+,16 NaGdF4:Yb3+, Tm3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and NaGdF4:Yb3+, Ho3+.18
17 and NaGdF4:Yb3+, Ho3+.18
Nowadays, many blue phosphors have been successfully prepared, such as KSrPO4:Eu2+,19 Ba3LaNa(PO4)3F:Eu2+20 and Na2Ba6(Si2O7)(SiO4)2:Ce3+.21 However, the emission from these phosphors is too blue to create a suitable white light. Tm3+ ions are usually used as efficient blue light emissive activators and have blue emissions which are mainly attributed to the 1D2 → 3F4 transition, which overlaps well with the 5I8 → 5G6 transition of Ho3+, as reported by Tian22 et al. and Xu23 et al. Whereas Ho3+ emits green light. Therefore, a change in hue from blue to green is expected in the Tm3+, Ho3+ co-doped system. For example, Silvestre24 presented the crystal growth of monoclinic KGd(WO4)2 co-doped with Tm3+ and Ho3+ by a top-seeded solution growth and slow cooling method. New types of fluorophosphate glass with different Tm3+, Ho3+ co-doping concentrations were prepared by Tian.22 In those chemical compounds, the Tm3+ and Ho3+ ions are excited by infrared rays. Up to now, studies on the luminescence properties of inorganic host materials co-doped with Tm3+ and Ho3+ ions under the excitation of UV light are limited. Following that, in this work, we aim to focus our attention on the luminescence of NaGdF4 co-doped Tm3+ and Ho3+ ions under the excitation of UV light.
Hence, in this work, the hexagonal phase NaGdF4:Tm3+, Ho3+ nanoparticles with down conversion luminescence including energy transfer and color tunable emissions have been reported and studied in detail.
2 Experimental
2.1 Materials
All the chemicals were used directly as received without further purification. Gd(NO3)3, Tm(NO3)3 and Ho(NO3)3 were prepared by dissolving the corresponding Gd2O3, Tm2O3 and Ho2O3 in HNO3 solution at an elevated temperature followed by evaporating superfluous HNO3. The sodium dodecyl sulfate (SDS) and sodium fluoride (NaF) were of analytical grade.
2.2 Preparation
A series of rare earth (RE)-doped NaGdF4 nanocrystals were synthesized by a facile hydrothermal process without further sintering treatment. In a typical procedure of preparing the representative NaGdF4:3%Tm3+, 0.5%Ho3+: 19.3 mL Gd(NO3)3, 1.2 mL Tm(NO3)3 and 0.2 mL Ho(NO3)3 were poured into a 100 mL flask with 1.1535 g (1:2 molar ratio for RE3+/SDS) of SDS. After vigorous stirring for 30 min, 1.007 g of NaF (1:12 molar ratio for RE(NO3)3/NaF) was slowly added into the above solution. After an additional agitation for 40 min, the resultant milky colloidal suspension was transferred to a Teflon bottle held in a stainless steel autoclave, and then heated at 180 °C for 24 h. Finally, after the autoclave was naturally cooled to room-temperature, the precipitate was separated by centrifugation, then washed sequentially with deionized water and ethanol several times, and then air dried at 60 °C for 12 h. Other samples were prepared with a similar procedure, except for changing the value of the RE(NO3)3.
2.3 Characterization
The morphology, size and composition of the samples were observed by a field emission electron microscope (FESEM) equipped with an energy-dispersive X-ray spectrometer (EDS).
X-ray powder diffraction (XRD) data for the prepared samples (NaGdF4:Tm3+, Ho3+) were taken using a Rigaku D/max-RA X-ray diffractometer with Cu Kα radiation (λ = 0.15406 nm) and a Ni filter, operating at 20 mA, 30 kV, and a scanning speed, step length and diffraction range of 10° min−1, 0.1° and 10–60°, respectively. The measuring of the photoluminescence (PL), photoluminescence excitation (PLE) spectra and the luminescence decay curves was performed by a HITACHI F-7000 Fluorescence Spectrophotometer using a 150 W Xe lamp as the excitation source and scanning at 1200 nm min−1. The excitation and emission slits were set to 2.5 and 5.0 nm. All of the measurements were performed at room temperature.
3 Results and discussion
3.1 Crystallization behaviour and morphology
Fig. 1 shows the representative XRD patterns of NaGdF4:3%Tm3+, x%Ho3+ with different doping content. One can see that no impurity peaks are observed in all of the samples. Simultaneously, all of the diffraction peaks are found to be in good agreement with those reported in JCPDS file 27-0699, regardless of the doping content, suggesting that the obtained samples are single phase and the doped Tm3+ or co-doped Tm3+, Ho3+ ions do not cause any significant changes in the host structure. Moreover, sharp and strong peaks indicate that the products synthesized at low temperature still have high crystallinity. The crystal structure of hexagonal NaGdF4 is shown in Fig. 1(b).25 As shown in Fig. 1(b), there are two crystallographic positions of the cations in the unit cell: six-fold coordinated Na+ sites and nine-fold coordinated Gd3+ sites. According to the effective ionic radii of cations, the rare-earth ions are proposed to occupy the Gd3+ sites or Na+ sites. This is because the effective radii of the Tm3+ ions (1.052 Å) and Ho3+ ions (1.072 Å) are similar to that of the Gd3+ ions (1.107 Å). In view of the effective radii, Tm3+ and Ho3+ ions are expected to occupy the Gd3+ sites preferentially. Simultaneously, based on the valence state analysis, the rare-earth ions are also much more likely to occupy the Gd3+ sites.
 |
| | Fig. 1 (a) XRD patterns of NaGdF4:3.0%Tm3+, x%Ho3+ (x = 0, 1.0, 1.5) samples. The standard for NaGdF4 (JCPDS card no. 27-0699) is shown as a reference. (b) Scheme of the hexagonal-phase NaGdF4 structures.25 | |
In order to study the influence of doped rare earth ions and surfactant on the morphology of the samples, FESEM measurements were done. The FESEM images shown in Fig. 2 present the morphology of the different samples prepared by the hydrothermal method. From Fig. 2, it can be seen that both the NaGdF4 (without SDS, (a)) and NaGdF4:3%Tm3+, 3%Ho3+ (without SDS, (b)) samples exhibit an irregular particle morphology with a diameter of about 50 nm, indicating that the co-doped Tm3+ and Ho3+ ions do not change the NaGdF4 morphology. The NaGdF4:3%Tm3+, 3%Ho3+ (with SDS, (c)) sample exhibits a rod-like morphology with a diameter of about 50 nm and a length of about 250 nm. Furthermore, the nanorod is assembled by the nanoparticles with a diameter of about 50 nm. This indicates that SDS plays an important role in the formation of nanorods. SDS is a kind of anionic surfactant. It enables a reduction in the surface tension of the solution and hinders the formation of a new phase in the process of crystal growth. In the formation of the nanorods, it can act as structural guide reagent, therefore, the nanoparticles self-assemble and aggregate into a rod-like shape. The EDS result of NaGdF4:3%Tm3+, 3.0% Ho3+ (Fig. 2(d)) reveals that the nanorods are mainly composed of Na, Gd, Tm, Ho and F. Note that the strong signals for Pt come from the platinum spraying process. All of these results further prove that the samples are NaGdF4:Tm3+, Ho3+. In the following work, all the luminescence properties of Tm3+ and/or Ho3+ doped NaGdF4 are measured using the samples prepared with SDS.
 |
| | Fig. 2 FESEM images of NaGdF4 (without SDS, (a)), NaGdF4:3%Tm3+, 3%Ho3+ (without SDS, (b)), and NaGdF4:3%Tm3+, 3%Ho3+ (with SDS, (c)). EDS spectrum of NaGdF4:3%Tm3+, 3%Ho3+ (d). | |
3.2 Luminescence properties of NaGdF4:Tm3+
Fig. 3 shows the PLE and PL spectra of NaGdF4:3%Tm3+. Monitoring the emission at 451 nm, the excitation spectrum displays an absorption band which is attributed to the transition of 3H6 → 1D2 at 358 nm. Upon excitation at 358 nm, the PL spectrum of NaGdF4:3%Tm3+ has a sharp emission peak which is due to the transition of 1D2 → 3F4 at 451 nm. The quenching concentration of NaGdF4:Tm3+ is 3.0% as shown in the inset of Fig. 3. Moreover, a bright blue light can be observed in the photo of NaGdF4:3%Tm3+ under excitation at 358 nm.
 |
| | Fig. 3 PLE and PL spectra of NaGdF4:3%Tm3+. Inset: the dependence of the emission intensity on Tm3+ concentration and a photo of NaGdF4:3%Tm3+ under excitation at 358 nm. | |
3.3 Luminescence properties of NaGdF4:Tm3+, Ho3+
The PLE spectra of NaGdF4:Tm3+, NaGdF4:Ho3+ and NaGdF4:Tm3+, Ho3+ nanoparticles at different emission wavelengths are exhibited in Fig. 4. The PLE spectra of NaGdF4:3%Tm3+ and NaGdF4:3%Tm3+, 0.5% Ho3+ which were monitored at the Tm3+ emission wavelength of 451 nm have the f–f transition of 3H6 → 1D2 of Tm3+ at 358 nm. The PLE spectra of NaGdF4:3%Ho3+ and NaGdF4:3%Tm3+, 0.5% Ho3+ which were monitored at the Ho3+ emission wavelength of 540 nm have 5I8 → 3H6 (359 nm), 5I8 → 5G5 (416 nm) and 5I8 → 5G6 (449 nm) transitions of Ho3+. Therefore, the excitation positions of the Tm3+ 3H6 → 1D2 (358 nm) and the Ho3+ 5I8 → 3H6 (359 nm) transitions are very close. In view of the synchronous emissions of these two ions, it is believed that a near-UV wavelength around 359 nm might be used to efficiently excite these co-doped nanoparticles, which properly fits the requirements for WLEDs.26 Moreover, in the NaGdF4:3%Tm3+, 0.5%Ho3+ phosphors, the excitation intensity of 359 nm monitored at the Ho3+ emission wavelength of 540 nm is weaker than that of the excitation peak of 358 nm when monitored at the Tm3+ emission wavelength of 451 nm. More importantly, the Tm3+ and Ho3+ co-doped NaGdF4 phosphors show a weak excitation peak (3H6 → 1D2, 358 nm) when compared with the only Tm3+ doped NaGdF4 phosphors, which indicates the expected energy transfer from Tm3+ to Ho3+. Meanwhile, the excitation peak overlap between 358 and 359 nm in the NaGdF4:3%Tm3+, 0.5%Ho3+ phosphors was seen when monitored at 540 nm. Therefore the energy transfer from Tm3+ to Ho3+ is still possible.
 |
| | Fig. 4 The excitation spectra of NaGdF4:Tm3+, NaGdF4:Ho3+ and NaGdF4:Tm3+, Ho3+ phosphors. | |
Fig. 5 displays the PL spectra of NaGdF4:3%Ho3+ (dashed line) and NaGdF4:3%Tm3+, 3%Ho3+ (solid line). From Fig. 5, the emissions at 485, 540 and 648 nm belong to the 5F3 → 5I8, 5S2 → 5I8 and 5F5 → 5I8 transitions of Ho3+, respectively. The emission at 451 nm belongs to the 1D2 → 3F4 transition of Tm3+. Moreover, the strong emission ascribed to the 5S2 → 5I8 (540 nm) transition of Ho3+ in NaGdF4:3%Tm3+, 3%Ho3+ (solid line), is about 2 times stronger than that of Ho3+ in NaGdF4:3%Ho3+ (dashed line), which suggests that the energy transfer occurred from Tm3+ to Ho3+.
 |
| | Fig. 5 The PL spectra of NaGdF4:3%Ho3+ (dashed line) and NaGdF4:3%Tm3+, 3%Ho3+ (solid line) under UV excitation (λex = 359 nm). | |
Fig. 6 shows the PL spectra of the NaGdF4:3%Tm3+, x%Ho3+ (x = 0, 0.5, 1.0, 1.5, 3.0) samples with different Ho3+ concentrations under excitation at 359 nm. From Fig. 6, one can see that all of the samples show a emission band in the blue region related to the electronic transitions of 1D2 → 3F4 (451 nm) of Tm3+.17 The emission bands in the green region are due to the 5F3 → 5I8 (485 nm) and 5S2 → 5I8 (540 nm) transitions of Ho3+.22,27 From the inset of Fig. 6, it is clear and reasonable to imply that the PL intensity of the Ho3+ activator (or energy acceptor) increases, whereas the PL intensity of the Tm3+ sensitizer (or energy donor) is simultaneously found to decrease monotonically, with an increasing concentration of Ho3+ ions, which indicates that there is an energy transfer from Tm3+ to Ho3+.
 |
| | Fig. 6 A series of PL spectra for NaGdF4:3%Tm3+, x%Ho3+(x = 0, 0.5, 1.0, 1.5, 3.0) under UV excitation (λex = 359 nm). Inset: the emission intensity of Tm3+ and Ho3+ with different Ho3+ doping content. | |
3.4 Energy transfer mechanism study
So as to make clear the possibility of the Tm3+ → Ho3+ energy transfer, the optical properties of single Tm3+, Ho3+-doped samples were studied. As described in Fig. 7, the comparison of the PL spectrum of NaGdF4:Tm3+ and the PLE spectrum of NaGdF4:Ho3+ discloses an obvious spectral overlap between the emission band of Tm3+ centered at 451 nm (1D2 → 3F4) and the excitation transaction of Ho3+ (5I8 → 5G6, 449 nm). Hence, an effective resonance-type energy transfer from Tm3+ to Ho3+ is expected.
 |
| | Fig. 7 Spectral overlap between PL spectrum of NaGdF4:3%Tm3+ (dashed line) and PLE spectrum of NaGdF4:3%Ho3+ (solid line). | |
The fluorescence decay process of the Tm3+ ions in NaGdF4:3%Tm3+, x%Ho3+ phosphors was investigated to further study the energy transfer between Tm3+ and Ho3+ by monitoring at 451 nm with an irradiation wavelength of 359 nm. From Fig. 8(a), one can see that all of the luminescence decay curves of Tm3+ in NaGdF4:Tm3+, Ho3+ can be fitted well with a single exponential function as
where
I and
I0 are the luminescence intensities at times
t and 0, respectively, and
τ is the luminescence life time. As shown in
Fig. 8(a), the corresponding luminescence decay times are determined to be 2.850, 2.447, 2.385 and 1.791 ms, respectively.
Fig. 8(b) clearly shows that the lifetimes for Tm
3+ ions are found to drastically decrease with increasing Ho
3+ concentration. This is because the energy absorbed by Tm
3+ transfers to Ho
3+, which is strong evidence for the energy transfer from Tm
3+ to Ho
3+, as reported by Shang.
28
 |
| | Fig. 8 (a) Decay curves for the luminescence of Tm3+ ions in NaGdF4:3%Tm3+, x%Ho3+ phosphors with different doped Ho3+ concentrations. (b and c) Dependence of the fluorescence lifetime of Tm3+ and the energy transfer efficiency ηT versus doped Ho3+ concentrations (x) in NaGdF4:3.0%Tm3+, x%Ho3+ phosphors. | |
The energy transfer efficiency (ηT) from the sensitizer Tm3+ ions to the activator Ho3+ ions can be calculated using the following formula29
where
ηT is the energy transfer efficiency and
I0 and
I are the peak intensities of the Tm
3+ ions in the absence and presence of Ho
3+ ions, respectively. Thus, the relationship between the energy-transfer efficiency and activator concentration of Ho
3+ ion can be obtained (
Fig. 8(c)). As shown in
Fig. 8(c), the energy transfer efficiency monotonically increases with an increase in Ho
3+ concentration and can even reach about 90% when
x = 3.0. The high energy transfer efficiency implies that the energy transfer from Tm
3+ to Ho
3+ ions is especially efficient. Simultaneously, the main reason for the increase in the Ho
3+ emission intensity is owing to the Tm
3+ → Ho
3+ energy transfer, rather than the energy absorbed by the Ho
3+ ions themselves.
7
3.5 Tunable color study
Aiming to research the effect of the Ho3+ ion doping content on the hue of NaGdF4:3%Tm3+, we synthesized a series of samples with the chemical compositions of NaGdF4:3%Tm3+, x%Ho3+ (x = 0, 0.5, 1.0, 1.5, 3.0). The Commission International de L’Eclairage (CIE) chromaticity coordinates of NaGdF4:NaGdF4:3%Tm3+, x%Ho3+ are presented in Fig. 9 and Table 1. The CIE chromaticity coordinate of pure Tm3+ activated NaGdF4 is (0.179, 0.097), corresponding to a purplish blue color. From the CIE diagram of NaGdF4:Tm3+, Ho3+ nanoparticles, it can be seen that with increasing concentration of Ho3+ ions, the chromaticity coordinates (x, y) can vary from (0.202, 0.194) to (0.219, 0.293), corresponding to the hue of these powders changing gradually from blue through to light blue and ultimately to bluish green. The digital photographs are also shown in the picture. The hues of the obtained samples change from purplish blue to light blue and eventually to bluish green as indicated in the inset of Fig. 9, which also demonstrates the role of the Ho3+ ions.
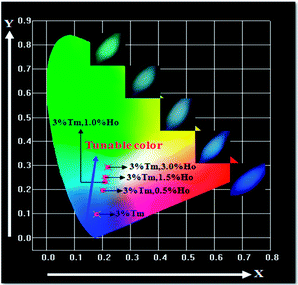 |
| | Fig. 9 CIE chromaticity diagram for NaGdF4:3%Tm3+, x%Ho3+ (x = 0, 0.5, 1.0, 1.5, 3.0) excited at 359 nm. | |
Table 1 The corresponding chromaticity coordinates of selected NaGdF4:Tm3+, Ho3+ nanoparticles
| Samples |
CIE (x, y) |
Color |
| NaGdF4:0.03Tm3+ |
(0.179, 0.097) |
Purplish blue |
| NaGdF4:0.03Tm3+, 0.005Ho3+ |
(0.202, 0.194) |
Blue |
| NaGdF4:0.03Tm3+, 0.10Ho3+ |
(0.211, 0.235) |
Light blue |
| NaGdF4:0.03Tm3+, 0.15Ho3+ |
(0.212, 0.248) |
Light blue |
| NaGdF4:0.03Tm3+, 0.30EHo3+ |
(0.219, 0.293) |
Bluish green |
4 Conclusions
In summary, a series of Tm3+, Ho3+ co-doped NaGdF4 nanoparticles were synthesized. The XRD patterns confirmed their hexagonal structure. In the NaGdF4:Tm3+, Ho3+ systems, the energy transfer efficiency increases with an increase in Ho3+ concentration. For the NaGdF4:3%Tm3+, x%Ho3+ (x = 0, 0.5, 1.0, 1.5, 3.0) nanoparticles, the emission could be tuned from purplish blue through to blue and ultimately to bluish green. Therefore, the β-NaGdF4 nanoparticles could have potential applications in fields such as color displays and solid-state lighting.
Acknowledgements
This work was supported by the National Science Foundation of P. R. China (NSFC) (Grant no. 51072026, 50972020) and the Development of Science and Technology plan projects of Jilin province (Grant no. 20130206002 GX).
Notes and references
- S. Gai, P. Yang, D. Wang, C. Li, N. Niu, F. He and X. Li, CrystEngComm, 2011, 13, 5480–5487 RSC.
- S. Huang, J. Xu, Z. Zhang, X. Zhang, L. Wang, S. Gai, F. He, N. Niu, M. Zhang and P. Yang, J. Mater. Chem., 2012, 22, 16136–16144 RSC.
- J. Li, J. Zhang, Z. Hao, X. Zhang, J. Zhao and Y. Luo, Appl. Phys. Lett., 2012, 101, 121905–121914 CrossRef PubMed.
- X. Zhang, J. Zhang and M. Gong, Opt. Mater., 2014, 36, 850–853 CrossRef CAS PubMed.
- T. Grzyb, A. Gruszeczka, R. J. Wiglusz and S. Lis, J. Mater. Chem. C, 2013, 1, 5410–5418 RSC.
- M. Jiao, N. Guo, W. Lu, Y. Jia, W. Lv, Q. Zhao, B. Shao and H. You, Dalton Trans., 2013, 42, 12395–12402 RSC.
- Y. Jia, W. Lu, N. Guo, W. Lu, Q. Zhao and H. You, Phys. Chem. Chem. Phys., 2013, 15, 6057–6062 RSC.
- M. Pokhrel, L. C. Mimun, B. Yust, G. A. Kumar, A. Dhanale, L. Tang and D. K. Sardar, Nanoscale, 2014, 6, 1667–1674 RSC.
- C. Gong, Q. Li, R. Liu, Y. Hou, J. Wang, X. Dong, B. Liu, X. Yang, Z. Yao, X. Tan, D. Li, J. Liu, Z. Chen, B. Zou, T. Cui and B. Liu, Phys. Chem. Chem. Phys., 2013, 15, 19925–19931 RSC.
- M. Pang, X. Zhai, J. Feng, S. Song, R. Deng, Z. Wang, S. Yao, X. Ge and H. Zhang, Dalton Trans., 2014, 43, 10202–10207 RSC.
- D. Yang, X. Kang, M. Shang, G. Li, C. peng, C. Li and J. Lin, Nanoscale, 2011, 3, 2589–2595 RSC.
- Z.-L. Wang, J. H. Hao and H. L. W. Chan, J. Mater. Chem., 2010, 20, 3178–3185 RSC.
- S. B. Wang, Q. Li, L. Z. Pei and Q.-F. Zhang, Mater. Charact., 2010, 61, 824–830 CrossRef CAS PubMed.
- Y. Wu, D. Yang, X. Kang, C. Li and J. Lin, Mater. Res. Bull., 2013, 48, 2843–2849 CrossRef CAS PubMed.
- H. Guan, G.-X. Liu, J. Wang, X. T. Dong and W. S. Yu, Dalton Trans., 2014, 43, 10801–10808 RSC.
- Q. Cheng, J. Sui and W. Cai, Nanoscale, 2012, 4, 779–784 RSC.
- F. He, N. Niu, L. Wang, J. Xu, Y. Wang, G. Yang, S. Gai and P. Yang, Dalton Trans., 2013, 42, 10019–10028 RSC.
- F. Li, C. Li, X. Liu, Y. Chen, T. Bai, L. Wang, Z. Shi and S. Feng, Chem.–Eur. J., 2012, 18, 11641–11646 CrossRef CAS PubMed.
- P. Dai, X. Zhang, L. Bian, S. Lu, Y. Liu and X. Wang, J. Mater. Chem. C, 2013, 1, 4570–4576 RSC.
- M. Jiao, N. Guo, W. Lü, Y. Jia, W. Lv, Q. Zhao, B. Shao and H. You, Inorg. Chem., 2013, 52, 10340–10346 CrossRef CAS PubMed.
- W. Lv, Y. Jia, Q. Zhao, M. Jiao, B. Shao, W. Lv and H. You, RSC Adv., 2014, 4, 14074–14080 RSC.
- Y. Tian, L. Y. Zhang, R. R. Xu, L. L. Hu and J. J. Zhang, Appl. Phys. B, 2010, 101, 861–867 CrossRef CAS PubMed.
- R. Xu, Y. Tian, L. Hu and J. Zhang, Appl. Phys. B, 2012, 108, 597–602 CrossRef CAS.
- O. Silvestre, M. C. Pujol, F. Güell, M. Aguiló, F. Díaz, A. Brenier and G. Boulon, Appl. Phys. B, 2007, 87, 111–117 CrossRef CAS PubMed.
- Y. Liu, D. Wang, J. Shi, Q. Peng and Y. Li, Angew. Chem., 2013, 125, 4462–4465 CrossRef PubMed.
- G. S. R. Raju, J. Y. Park, H. C. Jung, E. Pavitra, B. K. Moon, J. H. Jeong and J. H. Kim, J. Mater. Chem., 2011, 21, 6136–6139 RSC.
- W. Gao, H. Zheng, Q. Han, E. He and R. Wang, CrystEngComm, 2014, 6697–6706 RSC.
- M. Shang, D. Geng, X. Kang, D. Yang, Y. Zhang and J. Lin, Inorg. Chem., 2012, 51, 11106–11116 CrossRef CAS PubMed.
- G. Li, D. Geng, M. Shang, C. Peng, Z. Cheng and J. Lin, J. Mater. Chem., 2011, 21, 13334–13344 RSC.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 








