
Palladium-catalyzed direct ortho-sulfonylation of azobenzenes with arylsulfonyl chlorides via C–H activation†
Chengcai Xiaac,
Zhenjiang Weic,
Chao Shenb,
Jun Xub,
Yong Yanga,
Weike Su*a and
Pengfei Zhang*ab
aPharmacy College, Zhejiang University of Technology, Hangzhou 310014, China
bCollege of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 310036, China
cPharmacy College, Taishan Medical University, Tai'an 271016, China. E-mail: chxyzpf@hotmail.com; Fax: +86-571-28862867; Tel: +86-571-28862867
First published on 8th June 2015
Abstract
A highly efficient and practical procedure to direct ortho-sulfonylation of azobenzenes' C–H bond with arylsulfonyl chlorides has been developed. The method was applicable to both electron-rich and electron-deficient substrates and delivered good yields for 21 examples. This reaction provides a convenient access to ortho-sulfonylated azobenzenes under mild conditions.
Introduction
Transition-metal-catalyzed C–C and C–heteroatom bond formation via C–H bond activation has been broadly explored because it is an efficient method to construct heterocyclic compounds.1 Recently, more and more effort has been made to develop many techniques to control the reaction selectivity by assistance of a directing group. Among them, examples such as 2-aryloxypyridines,2 quinoline N-oxide,3 arylpyrazoles,4 triazene azoxybenzenes,5 quinoline,6 and 2-aryl-1,2,3-triazoles group,7 etc. were proved to be versatile directing groups to obtain high regioselectivity on the ortho-C(sp2)–H bond. In recent years, azobenzenes have worked as a directing group to accelerate the C–H activation/functionalization process and have attracted more attention. For example, many groups have developed a palladium-catalyzed regioselective C–H bond activation of azoarenes and related compounds with alcohols,8 toluene,9 aldehydes,10 and α-oxocarboxylic acids11 to synthesize ortho-acylazoarenes. But, Sun and co-workers found that they obtained ortho-alkoxyazoarenes when PhI(OAc)2 was added as the oxidant in this reaction.12 Also, Hao and Li's group have developed a highly efficient method to synthesize diverse cinnolines and isoquinolines through the rhodium-catalyzed oxidative C–H activation of azobenzenes and ketazines with alkynes.13 Similarly, azobenzene derivatives such as ortho-acyloxyazoarenes,14 ortho-sulfonamideazoarenes,15 and ortho-arylazoarenes16 have been synthesized by this method.Aryl sulfones have attracted considerable interest for they are essential components in medicinal chemistry,17 synthetic intermediates18 and advanced organic materials.19 The increasing applications of sulfones have stimulated investigations on development of efficient processes for the synthesis of these compounds. For example, Xu reported a method of palladium-catalyzed direct sulfonylation of 2-aryloxypyridines on the ortho-position of the benzene ring using 2-pyridyloxyl as the directing group and sulfonyl chlorides as sulfonylation reagents.20 Saidi has developed an efficient meta sulfonation of 2-phenylpyridines in the presence of ruthenium(II) complexes.21 Zhao disclosed a method of palladium-catalyzed direct sulfonylation of 2-arylpyridines on the ortho-position of the benzene ring using 2-arylpyridine as the directing group.22 Wu's group reported Pd(II)-catalyzed C–H sulfonylation of azobenzenes with arylsulfonyl chlorides using K2S2O8 as oxidant.23
As a part of our continuing efforts in C–H bond activation reactions, we have recently developed many methods to form C–S, and C–C bonds.24 Based on these findings, we develop a simple and efficient procedure for the synthesis of various ortho-sulfonylated azobenzenes via palladium-catalyzed direct cross-coupling of azobenzenes with arylsulfonyl chlorides.
Results and discussion
We initiated our investigation on the model reaction of azobenzene (1a) with p-tolylsulfonyl chloride (2a) to optimize the reaction parameters (Table 1). To our delight, the C2-sulfonylation took place in the presence of Pd(OAc)2 (10 mol%) and K2CO3 (2 equiv.) in DMSO under air for 12 h, affording compound 3a in 41% yield (entry 1, Table 1). Without catalyst, the reaction could not take place at all. Thus, PdCl2, [PdCl(allyl)]2, Pd(COD)Cl2, Pd(CH3CN)2Cl2, CuI and CuCl were tested to catalyze this reaction, in which Pd(CH3CN)2Cl2 gave the best result (entries 1–7, Table 1). K2CO3 was superior to other bases, such as Na2CO3, KOAc, NaOAc, CsCO3, NaHCO3 and KF (entries 8–13, Table 1). The solvent also played an important role in the reaction. Solvents such as DMF, NMP, CH3CN, 1,4-dioxane, toluene, and DMSO were screened, and 1,4-dioxane was found to be superior to the others (entries 5 and 14–18), affording 3a in 92% yield (entry 17, Table 1). The yield decreased to 73% when the catalyst loading was reduced to 5 mol% from 10 mol% (entry 19, Table 1). After surveying a variety of catalysts, bases, solvents, and catalyst loadings, we found that the combination of 10 mol% of Pd(CH3CN)2Cl2 and 2 equiv. of K2CO3 in 1,4-dioxane at 130 °C for 12 h served as the optimal conditions for this transformation. These results indicated that this transformation was facile and practical, as it did not require the use of strong bases, and the oxidants exclusion of air.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.6 equiv.), catalyst (10 mol%), base (2.0 equiv.), 4A MS (100 mg) and solvent (2.0 mL) under air at 130 °C for 12 h, unless otherwise noted.b Isolated yields.c Pd(CH3CN)2Cl2 (5 mol%). | ||||
| 1 | Pd(OAc)2 | K2CO3 | DMSO | 41 |
| 2 | PdCl2 | K2CO3 | DMSO | 33 |
| 3 | [PdCl(allyl)]2 | K2CO3 | DMSO | 39 |
| 4 | Pd(COD)Cl2 | K2CO3 | DMSO | 15 |
| 5 | Pd(CH3CN)2Cl2 | K2CO3 | DMSO | 77 |
| 6 | CuI | K2CO3 | DMSO | N.R. |
| 7 | CuCl | K2CO3 | DMSO | N.R. |
| 8 | Pd(CH3CN)2Cl2 | Na2CO3 | DMSO | 40 |
| 9 | Pd(CH3CN)2Cl2 | KOAc | DMSO | 31 |
| 10 | Pd(CH3CN)2Cl2 | NaOAc | DMSO | 35 |
| 11 | Pd(CH3CN)2Cl2 | Cs2CO3 | DMSO | 44 |
| 12 | Pd(CH3CN)2Cl2 | NaHCO3 | DMSO | 23 |
| 13 | Pd(CH3CN)2Cl2 | KF | DMSO | 31 |
| 14 | Pd(CH3CN)2Cl2 | K2CO3 | DMF | 45 |
| 15 | Pd(CH3CN)2Cl2 | K2CO3 | NMP | 67 |
| 16 | Pd(CH3CN)2Cl2 | K2CO3 | CH3CN | 73 |
| 17 | Pd(CH3CN)2Cl2 | K2CO3 | Dioxane | 92 |
| 18 | Pd(CH3CN)2Cl2 | K2CO3 | Toluene | 21 |
| 19 | Pd(CH3CN)2Cl2 | K2CO3 | Dioxan | 73c |

With the optimized reaction conditions in hand, the reactivities of different arylsulfonyl chlorides as the sulfonylation reagents were investigated. The results are revealed in Table 2, the C–H sulfonylation of azobenzene (1a) with arylsulfonyl chlorides could proceed smoothly and furnish the corresponding ortho-substituted products 3a–n in 63–92% yields (Table 2, entries 1–14). The substrates with a para-electron-donating group afforded the products 3a, 3g, 3n in excellent yields (92%, 84% and 91%). When arylsulfonyl chlorides were substituted at the para position with a electron-withdrawing group (such as 4-F, 4-Br, 4-methoxy, 4-Cl groups) there also afforded the products 3b, 3c, 3d and 3m in good yields (75–86%). But the substrates with a strong electron-withdrawing group (–NO2) on para and meta position provided the corresponding product 3d, 3f in low yields (67%, 63%). Heteroarysulfonyl chlorides, such as 3,5-dimethyl-isoxazole-4-sulfonyl chloride, thiophene-2-sulfonyl chloride, 5-chloro-3-methylbenzo[b]thiophene-2-sulfonyl chloride, and methyl 2-(chlorosulfonyl)thiophene-3-carboxylate were tested as the substrates. The corresponding ortho-sulfonylation products 3h, 3i, 3j, and 3k were obtained in good yields (73–90%).
|
|||
|---|---|---|---|
| Entry | Sulfinates | Product | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2 (0.6 equiv.), catalyst (10 mol%), base (2.0 equiv.), 4A MS (100 mg) and solvent (2.0 mL) under air at 130 °C for 12 h, unless otherwise noted.b Isolated yields. | |||
| 1 |  |
 |
92 |
| 2 |  |
 |
76 |
| 3 |  |
 |
86 |
| 4 |  |
 |
67 |
| 5 |  |
 |
75 |
| 6 |  |
 |
63 |
| 7 |  |
 |
84 |
| 8 |  |
 |
90 |
| 9 |  |
 |
87 |
| 10 |  |
 |
89 |
| 11 |  |
 |
73 |
| 12 |  |
 |
90 |
| 13 |  |
 |
82 |
| 14 |  |
 |
91 |

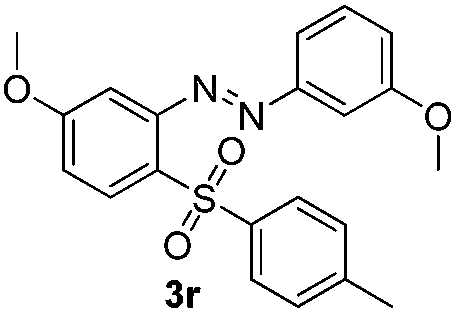
After screening of different arylsulfonyl chlorides, we explored the scope of differently substituted azobenzenes. Representative azoxybenzenes were firstly synthesized and examined. The results are shown in Table 3. It was found that these electron-rich azobenzenes gave higher yields than those electron-deficient azobenzenes. For instance, the reactions of 4,4′-dimethyl-azoxybenzene, 3,3′-dimethylazoxybenzene, 4,4′-di methoxy azoxybenzene, 3,3′-di methoxy azoxybenzene, provided the corresponding products in 75–86% yields (3o–3r). The reactions of unsymmetrical azobenzenes also proceeded smoothly and gave the products which could be determined by 1H NMR, were obtained in good yields (71% for 3t; 78% for 3u, respectively).













A single crystal of 3b was obtained from trichloromethane. Its crystal structure (Fig. 1) exhibited that such palladium-catalyzed direct sulfonylation of azobenzene on the ortho-position. The crystal data, refinement parameters, bond lengths and bond angles are given in the Table S1.†
 | ||
| Fig. 1 ORTEP diagram of catalyst 3b showing atom labelling scheme. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at 30% probability. | ||
In addition, when some radical scavengers such as TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) or HQ (hydroquinone) were used in this C–S coupling reaction, the reaction can be carried out smoothly. So this experiment can exclude the free radical mechanism. On the basis of these results and other previous related studies,19–22 a plausible reaction mechanism of this palladium-catalyzed sulfonylation of an azobenzene compounds is proposed, as shown in Scheme 1. Step (i), azobenzene 1a firstly reacts with Pd(CH3CN)2Cl2 to form a cyclopalladated(II) intermediate through ortho-C–H bond insertion. In step (ii), the sulfonylation could go through a direct displacement-type reaction to give the final product 3a. Meanwhile, the Pd(II) was regenerated for the next catalytic cycle.
 | ||
| Scheme 1 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed a palladium-catalyzed direct C(sp2)–H sulfonylation of azobenzene with arylsulfonyl Chlorides. The reaction exhibits a good tolerance for a broad range of general functional groups. The present work demonstrated the utility of azobenzene as a removable directing group through the direct C–H bond activation/functionalization and deprotecting group to form ortho-sulfonylated azobenzenes.Experimental section
General information
All reactions were run under argon in Schlenk tubes using vacuum lines. DMF, NMP, CH3CN, 1,4-dioxane, toluene, and DMSO, analytical grade were not distilled before use. Commercial arylsulfonyl chlorides and azobenzenes were used without purification. 1H NMR, 13C NMR spectra were recorded using a 500 MHz spectrometer in CDCl3 and DMSO with shifts referenced to SiMe4 (δ = 0). IR spectra were recorded on an FTIR spectrophotometer. Melting points were determined by using a local hot-stage melting point apparatus and are uncorrected. Elemental analyses were carried out on a CHN analyzer. Mass spectra were recorded using LC-MS and HRMS (ESI-TOF analyzer) equipment.General procedure for preparation of azoxybenzenes
All of the azo-compounds were prepared from arylamines, according to the literature.1 Mix CuBr (4.2 mg, 0.03 mmol), pyridine (8.7 mg, 0.09 mmol), arylamines (93 mg, 1 mmol) in toluene (4 mL) under air (1 atm). The reaction mixture was vigorously stirred at 60 °C for 20 h. After cooling down to room temperature and concentrating in vacuum, the residue was purified by flash chromatography on a short silica gel (eluent: petroleum ether) to afford azo-compound.General procedure for palladium-catalyzed direct ortho-sulfonylation of azobenzenes with arylsulfonyl chlorides via C–H activation
Mix azoic compound (0.5 equiv.), benzene sulfonyl chloride (0.6 equiv.), Pd(CH3CN)2Cl2 (10 mol%), K2CO3 (2 equiv.), 4A MS (100 mg) in 1,4-dioxane (2 mL) under air. The reaction mixture was vigorously stirred at 130 °C for 12 h. After cooling down to room temperature and concentrating in vacuum, the residue was purified by flash chromatography on a short silica gel to afford corresponding product.Diazene (1a)1
Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (no. LZ13B020001), National Natural Science Foundation of China (no. 21376058) and Major scientific and technological innovation projects of Hangzhou City (no. 20122511A43).Notes and references
- (a) G. Y. Song, F. Wang and X. W. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (b) Y. Fall, H. Doucet and M. Santelli, ChemSusChem, 2009, 2, 153–157 CrossRef CAS PubMed; (c) H. Y. Fu, L. Chen and H. Doucet, J. Org. Chem., 2012, 77, 4473–4478 CrossRef CAS PubMed; (d) A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 5206–5210 CrossRef CAS PubMed; (e) A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 1036–1041 CrossRef CAS PubMed; (f) H. Cao, Y. G. Lin, H. Y. Zhan, Z. D. Du, X. L. Lin, Q. M. Liang and H. Zhang, RSC Adv., 2012, 2, 5972–5975 RSC; (g) A. Nova, R. Mas-Balleste and A. Liedos, Organometallics, 2012, 31, 1245–1256 CrossRef CAS; (h) Y. S. Qiu, Y. Y. Kuang and J. Wu, Adv. Synth. Catal., 2014, 356(17), 3483–3504 CrossRef PubMed.
- (a) C. P. David, A. L. M. Geibel, E. M. N. J. Klein and R. Tobias, J. Am. Chem. Soc., 2009, 131, 17050–17051 CrossRef PubMed; (b) M. JoyRacowski, R. D. Allison and S. S. Melanie, J. Am. Chem. Soc., 2009, 131, 10974–10983 CrossRef PubMed.
- Z. Y. Wu, H. Y. Song, X. L. Cui, C. Pi, W. W. Du and Y. J. Wu, Org. Lett., 2013, 15(6), 1270–1273 CrossRef CAS PubMed.
- P. M. Liu and G. F. Christopher, Org. Lett., 2013, 15(22), 5862–5865 CrossRef CAS PubMed.
- H. Sun, C. M. Wang, Y. F. Yang, P. Chen, Y. D. Wu and X. H. Zhang, J. Org. Chem., 2014, 79(24), 11863–11872 CrossRef CAS PubMed.
- (a) R. D. Allison, L. H. Kami and S. S. Melanie, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef; (b) X. F. Cong and X. M. Zeng, Org. Lett., 2014, 16, 3716–3719 CrossRef CAS PubMed.
- S. P. Shi and C. X. Kuang, J. Org. Chem., 2014, 79, 6105–6112 CrossRef CAS PubMed.
- H. Tang, C. Qian, D. E. Lin, H. F. Jiang and W. Zeng, Adv. Synth. Catal., 2014, 356, 519–527 CrossRef CAS PubMed.
- H. Y. Song, D. Chen, C. Pi, X. L. Cui and Y. J. Wu, J. Org. Chem., 2014, 79, 2955–2962 CrossRef CAS PubMed.
- M. L. Yi, X. L. Cui, C. W. Zhu, C. Pi, W. M. Zhu and Y. Wu, Asian J. Org. Chem., 2015, 4, 38–41 CrossRef CAS PubMed.
- (a) H. J. Li, P. H. Li, Q. Zhao and L. Wang, Chem. Commun., 2013, 49, 9170–9172 RSC; (b) Z. Y. Li, D. D. Li and G. W. Wang, J. Org. Chem., 2013, 78, 10414–10420 CrossRef CAS PubMed.
- Z. W. Yin, X. Q. Jiang and P. P. Sun, J. Org. Chem., 2013, 78, 10002–10007 CrossRef CAS PubMed.
- (a) D. B. Zhao, Q. Wu, X. L. Huang, F. J. Song, T. Y. Lv and J. S. You, Chem. Eur. J, 2013, 19, 6239–6244 CrossRef CAS PubMed; (b) W. J. Han, G. Y. Zhang, G. X. Li and H. M. Huang, Org. Lett., 2014, 16, 3532–3535 CrossRef CAS PubMed; (c) M. Krishnamoorthy and C. H. Cheng, Chem.–Eur. J., 2013, 19, 6198–6202 CrossRef PubMed.
- C. Ian, D. E. Lin, Y. F. Deng, X. Q. Zhang, H. F. Jiang, G. Miao, X. H. Tang and W. Zeng, Org. Biomol. Chem., 2014, 12, 5866–5875 Search PubMed.
- X. F. Jia and J. Han, J. Org. Chem., 2014, 79, 4180–4185 CrossRef CAS PubMed.
- S. Miyamura, H. Tsurugi, T. Satoh and M. Miura, J. Organomet. Chem., 2008, 693, 2438–2442 CrossRef CAS PubMed.
- (a) L. Regina, A. Coluccia, A. Brancale, F. Piscitelli, V. Gatti, G. Maga, A. Samuele, C. Pannecouque, D. Schols, J. Balzarini, E. Novellino and R. Silvestri, J. Med. Chem., 2011, 54, 1587 CrossRef PubMed; (b) S. F. Barbuceanu, G. L. Almajan, I. Saramet, C. Draghici, A. I. Tarcomnicu and G. Bancescu, Eur. J. Med. Chem., 2009, 44, 4752 CrossRef CAS PubMed; (c) D. P. Becker, T. E. Barta, L. J. Bedell, T. L. Boehm, B. R. Bond, J. Carroll, C. P. Carron, G. A. Decrescenzo, A. M. Easton, J. N. Freskos, C. L. Funckes-Shippy, M. Heron, S. Hockerman, C. P. Howard, J. R. Kiefer, M. H. Li, K. J. Mathis, J. J. McDonald, P. P. Mehta, G. E. Munie, T. Sunyer, C. A. Swearingen, C. I. Villamil, D. Welsch, J. M. Williams, Y. Yu and J. Yao, J. Med. Chem., 2010, 53, 6653 CrossRef CAS PubMed; (d) E. Nuti, L. Panelli, F. Casalini, S. I. Avramova, E. Orlandini, S. Santamaria, S. Nencetti, T. Tuccinardi, A. Martinelli, G. Cercignani, N. D'Amelio, A. Maiocchi, F. Uggeri and A. Rossello, J. Med. Chem., 2009, 52, 6347 CrossRef CAS PubMed; (e) K. G. Liu, A. J. Robichaud, R. C. Bernotas, Y. Yan, J. R. Lo, M. Y. Zhang, Z. A. Hughes, C. Huselton, G. M. Zhang, J. Y. Zhang, D. M. Kowal, D. L. Smith, L. E. Schechter and T. A. Comery, J. Med. Chem., 2010, 53, 7639 CrossRef CAS PubMed; (f) A. V. Ivachtchenko, E. S. Golovina, M. G. Kadieva, V. M. Kysil, O. D. Mitkin, S. E. Tkachenko and I. M. Okun, J. Med. Chem., 2011, 54, 8161 CrossRef CAS PubMed; (g) S. Crosignani, A. Pretre, C. J. Lebrun, G. Fraboulet, J. Seenisamy, J. K. Augustine, M. Missotten, Y. Humbert, C. Cleva, N. Abla, H. Daff, O. Schott, M. Schneider, F. B. Charvillon, D. Rivron, I. Hamernig, J. F. Arrighi, M. Gaudet, S. C. Zimmerli, P. Juillard and Z. Johnson, J. Med. Chem., 2011, 54, 7299 CrossRef CAS PubMed.
- (a) F. Colobert, G. R. Ballesteros, F. R. Leroux, R. Ballesteros and B. Abarca, Tetrahedron Lett., 2007, 48, 6896 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (c) C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590 CrossRef CAS PubMed; (d) C. P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183 CrossRef CAS PubMed; (e) J. M. Khurana, V. Sharma and S. A. Chacko, Tetrahedron, 2007, 63, 966 CrossRef CAS PubMed; (f) A. Costa, C. Najera and J. M. Sansano, J. Org. Chem., 2002, 67, 5216 CrossRef CAS PubMed.
- (a) C. A. Zificsak and D. J. Hlasta, Tetrahedron, 2004, 60, 8991 CrossRef CAS PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (c) B. Ligault, I. Petrov, S. I. Gorelsky and K. Fagnou, J. Org. Chem., 2010, 75, 1047 CrossRef PubMed; (d) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- Y. F. Xu, P. Liu, S. L. Li and P. P. Sun, J. Org. Chem., 2015, 80, 1269–1274 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, K. K. Gabriele, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- X. D. Zhao, E. Dimitrijević and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466–3467 CrossRef CAS PubMed.
- D. Zhang, X. L. Cui, Q. Q. Zhang and Y. J. Wu, J. Org. Chem., 2015, 80, 1517–1522 CrossRef CAS PubMed.
- (a) C. Shen, H. J. Xia, H. Yan, X. Z. Chen, S. Ranjit, D. Tan, R. Lee, K. W. Huang, P. F. Zhang and X. G. Liu, Chem. Sci., 2012, 3, 2388–2393 RSC; (b) C. Shen, J. Xu, W. B. Yu and P. F. Zhang, Green Chem., 2014, 16, 3007–3012 RSC; (c) C. Shen, P. F. Zhang, Q. Sun, S. Q. Bai, T. S. A. Hor and X. G. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (d) C. Shen, H. Y. Shen, M. Yang, C. C. Xia and P. F. Zhang, Green Chem., 2015, 17, 225–230 RSC; (e) H. Y. Shen, C. Shen, A. M. Wang and P. F. Zhang, Catal. Sci. Technol., 2015, 5, 2065–2071 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, 13C NMR spectrum, GC/MS profile, HRMS profile. CCDC 1057250. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra06474k |
| This journal is © The Royal Society of Chemistry 2015 |