
Enantiomeric 3-arylcoumarins and 2-arylcoumarones from the roots of Glycyrrhiza uralensis as protein tyrosine phosphatase 1B (PTP1B) inhibitors†
Abstract
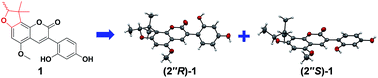
Glycyfuranocoumarin A–C (1–3), three 3-arylcoumarins, and glycyfuranocoumarone A (4), one 2-arylcoumarone, were isolated in the racemic form from the roots of Glycyrrhiza uralensis. Their structures were elucidated by NMR, 2D NMR and HRESIMS data analyses. All the structures contain a 2,3-dihydro-2,3,3-trimethylbenzofuran ring. Chiral HPLC was used to obtain the optically pure enantiomers, (2′′R)-1 and (2′′S)-1. Their absolute configurations were determined by X-ray crystallography. This is the first unambiguous determination of the absolute configuration of 2,3-dihydro-2,3,3-trimethylbenzofurans. (2′′R)-1 and (2′′S)-1, as well as 2–4, exhibited significant inhibitory activities against protein tyrosine phosphatase 1B (PTP1B). Among them, 4 showed an IC50 of 2.2 μM.
 Please wait while we load your content...
Please wait while we load your content...

