DOI:
10.1039/C5RA06449J
(Paper)
RSC Adv., 2015,
5, 42414-42421
Heterostructured nanocomposite tin phthalocyanine@mesoporous ceria (SnPc@CeO2) for photoreduction of CO2 in visible light†
Received
17th April 2015
, Accepted 5th May 2015
First published on 5th May 2015
Abstract
Heterostructured tin phthalocyanine supported to mesoporous ceria was synthesized and used a photocatalyst for CO2 reduction under visible light. The photoreduction CO2 activities of the heterostructures were investigated in the presence of triethylamine as sacrificial agent. The developed photocatalyst exhibited high catalytic activity for photoreduction of CO2 and after 24 hours of visible light irradiation 2342 μmol g−1 cat of methanol (ϕMeOH = 0.0223 or 2.23%) and 840 μmol g−1 cat of CO (ϕCO = 0.0026 or 0.26%) were obtained as the major reaction products. The methanol formation rate (RMeOH) and CO formation rate (RCO) was found to be 97.5 μmol h−1 g−1 cat and 35.0 μmol h−1 g−1 cat respectively. While under the identical experimental conditions mesoporous ceria (meso-CeO2) gave only 316 μmol g−1 cat of methanol (ϕMeOH = 0.003 or 0.30%) and 126 μmol g−1 cat CO (ϕCO = 0.0004 or 0.04%) with product formation rate RMeOH = 13.2 μmol h−1 g−1 cat and RCO = 5.3 μmol h−1 g−1 cat. Furthermore, the recovered catalyst showed consistent catalytic activity for at least five runs without any significant loss in product yields.
1 Introduction
Continuous rise in emission of anthropological carbon dioxide (CO2) and the depletion of fossil resources have driven the research interest in developing processes for converting CO2 to value added chemicals.1 Among the different processes known, solar assisted photocatalytic conversion of CO2 to renewable fuels is regarded as a long term beneficial approaches that not only tackles global warming but also partly fulfils energy demands.2 In this regard, Inoue et al.3 in 1979 first reported photoreduction of CO2 to organic compounds under UV light and after that increasing efforts are being focused on extending the light response of TiO2 to visible region.4 However, only limited successes were achieved due to the wide band gap of the matrix TiO2 (3.0–3.2 eV). Recently, several semiconductor based photocatalysts such as modified TiO2,5 CdS,6 ZnS,7 mixed metal oxides8 for CO2 reduction have been developed, but the efficiencies are still far from practical applications because of fast charge recombination rates. To overcome the limitations of the narrow visible-light absorption and fast charge recombination of semiconductor photocatalysts, several strategies for instance band-gap engineering by doping of various metals (Cu,9 Ag,10 Ru11 etc.) and by attaching visible light absorbing molecular complexes12 on photoactive supports to give heterostructured materials have recently been developed. Particularly, construction of heterostructured materials by heterogenization of homogeneous metal complexes is fascinating as it provides high efficiencies as compared to semiconductors as well as facile recovery and recycling ability of these catalysts make them more attractive from economical viewpoints. Among the various transition metal based complexes, polypyridyl complexes of ruthenium,13 iridium14 have been used as photocatalyst for photoreduction of CO2 under visible light. However, expensive nature and limited accessibility of these precious metal catalysts make the developed processed less desirable from economical view points.
Phthalocyanines and porphyrins are important class of organic dyes which posses a number of unique properties such as excellent semi-conductivity, photoconductivity, chemical stability and optical absorption in the UV-Vis region.15 Particularly, metallophthalocyanines owing to their ease of synthesis, high thermal stability, excellent electrical and optical properties have attracted a great deal of attention and have been used as light-harvesting materials to broaden the absorption range of semiconductors.16 Anchoring of these dye complex units to the nanocrystalline semiconductor supports enable ultrafast injection of electrons from the excited state into the conduction band of semiconductor to give improved conversion efficiency. In this regard, recently we reported graphene oxide (GO)–cobalt phthalocyanine complex as an effective, recyclable visible light photocatalyst to photoreduce CO2 to methanol.17 In continuation to our on-going research on visible light assisted photoreduction of CO2,18 herein we report an efficient heterostructured composite constructed by the combination of tin phthalocyanine and mesoporous ceria for the photoreduction of carbon dioxide using triethylamine as sacrificial donor under visible light irradiation.
2 Results and discussion
2.1 Synthesis and characterization of catalyst
During the present study, meso-CeO2 was prepared by combustion method using chitosan as template as suggested in the literature.19 The synthesized meso-CeO2 was first treated by water to enhance the –OH groups on the surface. Tin(IV) phthalocyanine dichloride was attached to meso-CeO2 through axial position by taking advantage of –OH functional groups located on the surface of meso-CeO2 (Scheme 1).
 |
| | Scheme 1 Synthesis of SnPc@CeO2 catalyst. | |
Fig. 1 shows the SEM images of uncalcined ceria, meso-CeO2 and supported SnPc@CeO2. The SEM image of uncalcined CeO2 samples show unorganised rough surface (Fig. 1a). The SEM image of meso-CeO2 show erupted ridge type structures that may be appeared due to the burning of template in the combustion step (Fig. 1b). The supported catalyst i.e. SnPc@CeO2 also showed similar crinkled morphology as of meso-CeO2 Fig. 1c. Furthermore appearance of Sn, Ce, C, N and O elements in EDX pattern of SnPc@CeO2 confirmed the presence of tin phthalocyanine in the synthesized material.
 |
| | Fig. 1 SEM images of (a) uncalcined CeO2 (b) meso-CeO2 and (c) SnPc@CeO2 and EDX pattern of (d) uncalcined CeO2 (e) meso-CeO2 and (f) SnPc@CeO2. | |
The microstructure of meso-CeO2 and SnPc@CeO2 was determined with TEM as shown in Fig. 2a and b, respectively. TEM image clearly indicated that the size of particles in the synthesized meso-CeO2 was in the range of 5–10 nm diameters (Fig. 2a). After the attachment of SnPc to meso-CeO2 although the morphology remained almost same with 5–10 nm diameter, however some aggregation was observed due to cementing with SnPc (Fig. 2b). SAED pattern of SnPc@CeO2 clearly depicted rings due to diffraction of 111, 220 and 311 plane with 1.65 Å, 1.05 Å and 0.82 Å d spacing of meso-CeO2 which explained the crystalline morphology of material (Fig. 2c). Further absence of sharp spot defines polycrystalline structure of material because of rapid heating during combustion process prevents large crystal growth.
 |
| | Fig. 2 TEM images of (a) meso-CeO2 (b) SnPc@CeO2 and (c) SAED pattern of SnPc@CeO2. | |
FTIR spectra of SnPcCl2 showed characteristics peaks due to phthalocyanine ring vibration at 723 cm−1 and 781 cm−1. The peaks at 1124 cm−1, 1581 cm−1 and 1656 cm−1 were assumed due to the aromatic ring vibrations, C–H bending vibrations, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibration and pyrrole ring vibrations of the phthalocyanine ring (Fig. 3a).20 FTIR spectra of meso-CeO2 showed its characteristic peaks at 1357 cm−1 and 1544 cm−1 (Fig. 3b). The peak at 1631 cm−1 and 3428 cm−1 were observed due to the adsorbed water on the surface.21 The presence of peaks of SnPc at 711 cm−1, 782 cm−1, 1118 cm−1, 1344 cm−1, 1463 cm−1 and 1583 cm−1 in heterostructured materials i.e. SnPc@CeO2 confirmed the successful attachment of tin(IV) phthalocyanine to meso-ceria support (Fig. 3c).
N vibration and pyrrole ring vibrations of the phthalocyanine ring (Fig. 3a).20 FTIR spectra of meso-CeO2 showed its characteristic peaks at 1357 cm−1 and 1544 cm−1 (Fig. 3b). The peak at 1631 cm−1 and 3428 cm−1 were observed due to the adsorbed water on the surface.21 The presence of peaks of SnPc at 711 cm−1, 782 cm−1, 1118 cm−1, 1344 cm−1, 1463 cm−1 and 1583 cm−1 in heterostructured materials i.e. SnPc@CeO2 confirmed the successful attachment of tin(IV) phthalocyanine to meso-ceria support (Fig. 3c).
 |
| | Fig. 3 FT-IR Spectra of (a) SnPcCl2 (b) meso-CeO2 (c) SnPc@CeO2. | |
Fig. 4 shows X-ray diffraction pattern of meso-CeO2 and SnPc@CeO2. The XRD diffraction pattern of meso-CeO2 shows its characteristics peaks at the 2θ value 28.4° (111), 33.5° (200), 47.5° (220), 56.2° (311), 69.8° (400) and 76.4° (331) which can be indexed to fcc cubic space group Fm3m (225) structure and was in good agreement with JCPDS card no. 34-0394 (Fig. 4a).22 Immobilization of tin phthalocyanine to meso-CeO2 in SnPc@CeO2 catalyst did not change diffraction pattern of meso-CeO2 which was most likely due to the lower loading (Fig. 4b).
 |
| | Fig. 4 XRD diffraction pattern of (a) meso-CeO2 (b) SnPc@CeO2. | |
Nitrogen adsorption–desorption isotherm was used to determine surface properties such as BET surface area (SBET), mean pore diameter (rp) and total pore volume (Vp). For the meso-CeO2 and SnPc@CeO2 the type (IV) loop of N2 adsorption–desorption isotherm was observed which suggested the mesoporous nature of materials according to IUPAC recommendation (Fig. S1†).23 The BET surface area (SBET) of meso-CeO2 (75.92 ± 2% m2 g−1) was significantly decreased in SnPc@CeO2 (20.52 ± 2% m2 g−1) which is due to the attachment of SnPc to the meso-CeO2 surface.
UV-Vis spectrum of tin(IV) phthalocyanine dichloride (SnPcCl2) collected in DMF, showed its characteristics absorption band at 298 nm (Soret band) and 688 nm (Q band) respectively due to macrocyclic π → π* ring transitions (Fig. 5a).24 The broad Q band of tin(IV) phthalocyanine was observed because tin(IV) phthalocyanine exist in aggregated form in solution and thus, the absorption band was originated due to the combined absorption of monomeric and aggregated forms.25 The UV-Vis spectrum of meso-CeO2 exhibited a strong absorption band in the UV region due to O-2p to Ce-4f transition (Fig. 5b).26 After the immobilization of SnPc to meso-CeO2, a relatively sharp Q band of tin(IV) phthalocyanine was appeared. This is most likely due to the formation of monomeric form of SnPc during the immobilization to meso-CeO2 support. The appearance of sharp Q band in UV-Vis spectra of SnPc@CeO2 was a strong indication of successful attachment of SnPc to meso-ceria.27 The increased visible light absorption profile indicates that the developed catalyst can work efficiently under visible light irradiation (Fig. 5c).
 |
| | Fig. 5 UV/Vis absorption spectra of (a) SnPcCl2 (b) meso-CeO2 (c) SnPc@CeO2. | |
Thermal stability of materials was determined by thermo gravimetric analysis as shown in Fig. 6. TGA thermogram of SnPcCl2 shows a very small weight loss between 150–180 °C due to degradation of residual solvent molecules and 1,2-dicyanobenzene used for the synthesis of SnPc (Fig. 6a). The sharp weight loss at around 535 °C was observed due to the degradation of phthalocyanine ring structure.28 For CeO2, first weight loss around at 100 °C was due to the evaporation of water and then a steady weight loss was observed (Fig. 6b).29 Heterostructured SnPc@CeO2 catalyst showed the first a very small weight loss at 50–100 °C due to moisture and second major weight loss between 350–575 °C due to degradation of immobilized SnPc and oxy-functionalities presented on the support (Fig. 6c).
 |
| | Fig. 6 DT-TGA thermogram of (a) SnPcCl2 (b) meso-CeO2 (c) SnPc@CeO2. | |
2.2 Photo-catalytic activation of CO2
The photocatalytic CO2 activation experiments were carried out by using meso-CeO2, SnPc@CeO2, and homogeneous SnPcCl2 in DMF and water and triethylamine mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). The reaction mixture was irradiated with 20 watt white cold LED flood light. To monitor the progress of reaction, liquid samples were withdrawn during the reaction and 1 μL sample was injected in GC-FID and gaseous products were analyzed with GC-TCD and GC-FID (using RGA column). Further a calibration curve was plotted by injecting calculated amount of methanol in GC-FID. Methanol was observed as major liquid reaction product so its yield was used for evaluating the performance of catalyst (Fig. S2†). A plot of methanol yield during time by using meso-CeO2, equimolar SnPcCl2 to SnPc@CeO2 catalyst and SnPc@CeO2 catalyst and was plotted in Fig. 7. After 24 hours of visible light irradiation the yield of methanol by using meso-CeO2, SnPcCl2 in equimolar to SnPc@CeO2 and SnPc@mesoCeO2 was found to be 316 μmol g−1-cat, 1402 μmol g−1 cat and 2342 μmol g−1 cat respectively. The corresponding methanol formation rate RMeOH by using meso-CeO2, homogeneous SnPcCl2 in equimolar to SnPc@CeO2 and SnPc@mesoCeO2 was calculated 13.2 μmol h−1 g−1 cat, 58.4 μmol h−1 g−1 cat and 97.5 μmol h−1 g−1 cat with quantum yield for methanol (ϕMeOH) 0.003 (0.30%), 0.0133 (1.33%) and 0.0223 (2.23%) respectively. The quantum yield for product was determined as follows:
:1:1). The reaction mixture was irradiated with 20 watt white cold LED flood light. To monitor the progress of reaction, liquid samples were withdrawn during the reaction and 1 μL sample was injected in GC-FID and gaseous products were analyzed with GC-TCD and GC-FID (using RGA column). Further a calibration curve was plotted by injecting calculated amount of methanol in GC-FID. Methanol was observed as major liquid reaction product so its yield was used for evaluating the performance of catalyst (Fig. S2†). A plot of methanol yield during time by using meso-CeO2, equimolar SnPcCl2 to SnPc@CeO2 catalyst and SnPc@CeO2 catalyst and was plotted in Fig. 7. After 24 hours of visible light irradiation the yield of methanol by using meso-CeO2, SnPcCl2 in equimolar to SnPc@CeO2 and SnPc@mesoCeO2 was found to be 316 μmol g−1-cat, 1402 μmol g−1 cat and 2342 μmol g−1 cat respectively. The corresponding methanol formation rate RMeOH by using meso-CeO2, homogeneous SnPcCl2 in equimolar to SnPc@CeO2 and SnPc@mesoCeO2 was calculated 13.2 μmol h−1 g−1 cat, 58.4 μmol h−1 g−1 cat and 97.5 μmol h−1 g−1 cat with quantum yield for methanol (ϕMeOH) 0.003 (0.30%), 0.0133 (1.33%) and 0.0223 (2.23%) respectively. The quantum yield for product was determined as follows:
| Quantum yield of product (ϕproduct) = [number of moles of product] × number of electrons required for reduction/[moles of incident photons] |
 |
| | Fig. 7 Photocatalytic methanol formation versus time by using (a) blank reaction (b) meso-CeO2 and (c) SnPcCl2 equimolar amount as in SnPc@CeO2 and (d) SnPc@CeO2. | |
For methanol and CO formation, number of electrons required is, respectively, 6 and 2.
The analysis of gaseous products after 24 h gave carbon monoxide as major reaction product along with the small amounts of hydrogen and oxygen. The yield of carbon monoxide by using meso-CeO2, homogenous SnPcCl2 and SnPc@CeO2 was found to be 126 μmol g−1 cat, 514 μmol g−1-cat and 840 μmol g−1 cat respectively. The carbon monoxide formation rate (RCO) by using meso-CeO2, homogenous SnPcCl2 and SnPc@CeO2 was found to be 5.3 μmol h−1 g−1 cat, 21.4 μmol h−1 g−1 cat, 35.0 μmol h−1 g−1 cat with the quantum yield 0.0004 (0.04%), 0.0016 (0.16%) and 0.0026 (0.26%) respectively. Hydrogen was observed only in the case of SnPc@CeO2, whereas it was found to be absent in case of using other catalyst components like CeO2 and SnPcCl2. The yield of hydrogen with SnPc@CeO2 catalyst was found to be 13.5 μmol g−1 cat (RH2 = 0.56 μmol h−1 g−1 cat) with 4.28 × 10−5 quantum yield. The catalytic selectivity for methanol by using meso-CeO2, SnPcCl2 and SnPc@CeO2 was calculated as 71%, 73% and 73% respectively.
Production of methanol with time under blank condition as well as using different catalyst components like meso-CeO2, homogeneous SnPcCl2 and heterogeneous SnPc@CeO2 is shown in Fig. 7. It is clear from the plot that SnPc@CeO2 gave highest product yield in comparison to SnPcCl2 and meso-CeO2. Furthermore, the lower product yield with homogeneous SnPcCl2 as compared to heterogeneous SnPc@CeO2 suggested the promoting role of ceria support in enhancing the photoreduction reaction. As shown in Fig. 7, the shape of the highest curve (SnPc@CeO2) typically looks like a saturation curve, i.e. is not linear. This is most likely due to the following reason: as we performed the photoreduction experiments in a closed vessel, so after the consumption of available CO2 a saturation curve is reached (Fig. 7). In order to confirm the stability, after 24 h of illumination we again re-purged the reaction mixture with CO2 and continued the reaction for additional 24 h by withdrawing the samples after every 4 h. After re-purging the solution with CO2, the methanol yield was found to be increased as similar as in first run and then approaching to the saturation point with the consumption of available CO2 (Fig. S4†).
The origin of methanol as a result of photoreduction product of carbon dioxide was ensured with three blank reactions. The first reaction was carried out in the absence of catalyst, second in the absence of light and the third by purging nitrogen in place of CO2 under otherwise identical conditions. There were no product was observed after a long period of exposure.
Further we have checked recyclability of catalyst. After the reaction SnPc@CeO2 catalyst was separated by centrifugation and washed with ethanol and dried. This catalyst was used for further reactions. After five recycling there was no significant loss in activity was observed (Fig. 8). The tin metal content in the recovered catalyst after five runs was found to be 0.34 wt% that was comparable to fresh catalyst 0.38 wt%. Furthermore to prove the heterogeneous nature of catalyst we added 100 mg catalyst in 10 mL water and then stirred it for 24 h. The catalyst was separated with centrifugation and the resulting filtrate was analysed by ICP-AES. The tin content in the filtrate was found to be 0.02 wt% which revealed that a marginal leaching had occurred during the reaction. These results suggested the higher stability as well as heterogeneous nature of the developed heterostructured photocatalyst without giving any significant leaching.
 |
| | Fig. 8 Reuse experiments of photocatalytic CO2 reduction by using SnPc@CeO2 catalyst. | |
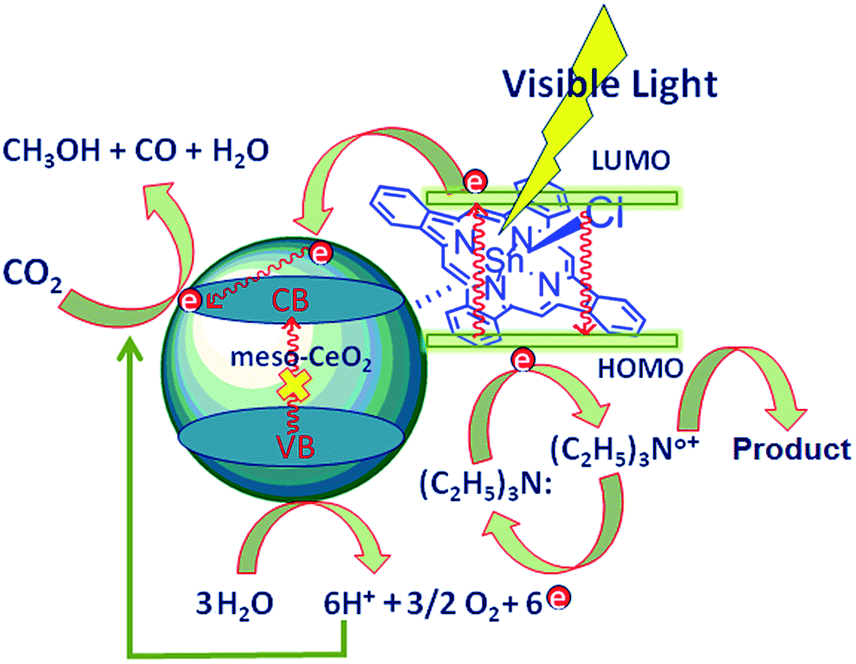
The better photocatalytic activity of SnPc@CeO2 towards the CO2 reduction was explained due to the better charge injection in the conduction band of CeO2. As mentioned in literature meso-ceria can absorb only in the UV region due to its high band gap.30 Tin(IV) phthalocyanine has good visible light absorption capacity with longer life time of its excited species as compared to ruthenium bipyridyl complexes and other phthalocyanine.31 So after absorption of the visible light SnPc excited to singlet (S1) excited state and transfer electrons to the conduction band of meso-CeO2.32 Due to continuous pumping of electrons by SnPc in the conduction band of CeO2 multiple electron transfer necessary for methanol formation is possible.17,18 These electrons in the conduction band of CeO2 were used for the reduction of CO2 adsorbed on the surface of CeO2. Triethylamine was used as a sacrificial donor to provide necessary electrons for the photoreduction process. After providing electrons triethylamine was converted to its degradation products.33,2f However, water provided necessary protons for the reaction as shown in the Scheme 2.34
| SnPc + hν → SnPc*(S1) (excited singlet state) |
| SnPc*(S1) + CeO2 → SnPco+ + 6e− (CeO2 conduction band) |
| 6e− (CeO2 conduction band) + CO2 + 6H+ → CH3OH + H2O |
| SnPco+ + TEA → SnPc + TEAo+ |
| TEAo+ → degradation products + e− (transferred to SnPc) |
 |
| | Scheme 2 Plausible mechanism of CO2 reduction over SnPc@CeO2 catalyst. | |
3 Conclusions
In summary, heterostructured SnPc@CeO2 photocatalyst was successfully achieved by the immobilization of tin(IV) phthalocyanine to mesoporous ceria through axial position by taking advantage of hydroxyl groups presented on the surface of meso-CeO2. The synthesized photocatalyst exhibited good photocatalytic activity under visible light irradiation and afforded methanol and carbon monoxide as major products. The developed catalyst was found to be more efficient as compared to the meso-CeO2 as well as homogeneous SnPcCl2 with the added benefits of facile recovery and consistent activity for recycling runs. With the use of SnPc@CeO2, the yield of methanol and CO after 24 h was attained as 2342 μmol g−1 cat and 840 μmol g−1 cat with quantum yields (ϕMeOH = 0.0223) and (ϕCO = 0.0026) respectively. The attained product formation rate for methanol (RMeOH) and CO (RCO) by using SnPc@CeO2 was 97.5 μmol h−1 g−1 cat and 35.0 μmol h−1 g−1 cat. The superior photocatalytic activity of the heterostructured catalyst was defined on the basis of better charge injection in the conduction band of meso-CeO2. We believe that our findings will be helpful for scientific community to develop low cost visible light active photocatalytic materials for conversion of CO2 to value added chemicals.
4 Experimental section
4.1 Materials
Tin(II) chloride dihydrate (98%), phthalonitrile (≥96.0%), cerium nitrate (99.99%) and the template chitosan (85% deacylated) was purchased from Alfa Aesar. Chloro naphthalene (≥85%) was purchased from Sigma-Aldrich. DMF and water used were of HPLC grade and procured from Merck. All other chemicals were of analytical grade and used without any further purification.
4.2 Techniques used
The rough surface morphology of materials were determined with the help of Field Emission Scanning Electron Microscopy (FE-SEM) performed on FE-SEM (Jeol Model JSM-6340F). Transmission Electron Microscopy (TEM) was used for the determination of ultrafine structure of materials and performed on FEI-TecnaiG2 TwinTEM operating at an acceleration voltage of 200 kV. TEM images were collected by depositing very dilute aqueous suspension of sample on carbon coated TEM grid. Vibrational spectra (FTIR) for identification of functionalities present in samples were collected on Perkin-Elmer spectrum RX-1 IR spectrophotometer using potassium bromide window. UV-Vis absorption spectra of SnPcCl2 in DMF and solid UV-Vis of other samples were collected on Perkin Elmer lambda-19 UV-Vis-NIR spectrophotometer using a 10 mm quartz cell, using BaSO4 as reference. To executing the phase structure and crystalline nature of material and powder X-ray diffractogram was recorded on Bruker D8 Advance diffractometer at 40 kV and 40 mA with Cu Kα radiation (λ = 0.15418 nm). Before analysis samples was prepared in glass slides and dried properly. Brunauer–Emmett–Teller (BET) surface area, Barret–Joiner–Halenda (BJH) porosity, pore volume etc. surface properties of samples were examined by N2 adsorption–desorption isotherm at 77 K by using VP; Micromeritics ASAP 2010. To obtaining thermal stability data and rough idea of chemical moiety presents in materials Thermo Gravimetric Analyses (TGA) was performed by using a thermal analyzer TA-SDT Q-600. Analysis was carried out in the temperature range of 40 to 800 °C under nitrogen flow with heating rate 10 °C min−1 metal content of SnPc@CeO2 catalyst was determine by ICP-AES analysis that was carried out by using Inductively Coupled Plasma Atomic Emission Spectrometer (ICP-AES, DRE, PS-3000UV, Leeman Labs Inc, USA). Elemental analysis was done for confirming the intact ring structure of phthalocyanine and no metal was leached out on catalyst. For illumination LED light (Model: HP-FL-20W-F, Hope LED Opto-Electric Co., Ltd) was used having following specification – Chip: KWE, Bridgelux, Epistar COB LED, 20 W, lum. output: 80–90l m W−1, beam angle: 120 degree and wavelength λ > 400 (maximum at 515 nm). Fig. S5† shows spectral power distribution (spectrum profile) for different wavelength.
4.3 Synthesis of mesoporous-ceria (meso-CeO2)19
For the synthesis of mesoporous CeO2 literature procedure using a modified template method was followed. In a 100 mL of 5% acetic acid solution, 3 g chitosan powder was dissolved with stirring for about 1 h. An aqueous solution of cerium nitrate (1.5 g) was added to this solution and stirred for 2 h. The obtained precursor was precipitated by adding 50% aqueous ammonia solution. Precipitate was dried at 60 °C and calcined at 550 °C for 5 hours and grinded.
4.4 Immobilization of tin(IV) phthalocyanine dichloride (SnPcCl2) to meso-CeO2 (SnPc@CeO2)
Tin(IV) phthalocyanine dichloride (SnPcCl2) was synthesized by following the literature method.35 Mesoporous ceria was first treated with water for 24 h to increase the –OH functionalities on the surface and then separated by centrifuge, dried at 60 °C. The immobilization of tin(IV) phthalocyanine dichloride to meso-CeO2 was carried out by refluxing SnPcCl2 (0.25 g) and meso-CeO2 (2.0 g) in ethanol for 12 hours in the presence of triethylamine. After cooling SnPc@CeO2 was separated by centrifuge and washed thoroughly with water, ethanol and dried at 70 °C under vacuum for 12 h. The Sn-metal content in the synthesized sample was determined by ICP-AES and was found to be 0.38 wt% (53 μmol SnPcCl2 g−1 cat).
4.5 Photocatalytic CO2 activation experiment
All the synthesized materials i.e. meso-CeO2, SnPc@CeO2 and homogeneous SnPcCl2 were tested for photocatalytic CO2 reduction using triethylamine as sacrificial donor. In a borosil vessel (5 cm dia.) water (10 mL), triethylamine (10 mL), and DMF (30 mL) was added and nitrogen was purged through this solution for 15 min to replace other dissolved gaseous. After that carbon dioxide was purged through this solution for 30 minutes for saturating the solution with carbon dioxide. The vessel was charged with 100 mg of catalyst and sealed with a rubber septum and irradiated under visible light by using 20 watt LED (Model no. HP-FL-20W-F, Hope LED Opto-Electric Co., Ltd) with stirring. To preventing from outer interference whole reaction setup was placed in a box. The intensity of light on vessel's surface was found 85 W m−2 measures with the help of intensity meter. Samples were withdrawn after every 2 hours interval with the help of a long needle and catalyst was removed by syringe filter (PTFE, 0.2 μm, 13 mm dia.). The obtained samples were injected in GC-FID (Varian CP-3800 by using 30 m long Stabilwax® w/Integra-Guard® column, flow rate: 0.5 mL min−1, injector temperature: 250 °C, FID detector temperature: 275 °C) for determination of liquid products. A calibration curve was plotted by injecting various concentration of methanol in GC-FID for quantification and to checking linear response and sensitivity to various concentrations. Gaseous products were determined with the help of GC-TCD and GC-FID (Agilent 7890A GC system) using capillary column (RGA, refinery gas analyzer) at the flow rate (H2: 35 mL min−1, air: 350 mL min−1, makeup flow: 27 mL min−1, for TCD reference flow: 45 mL min−1, helium flow: 2 mL min−1), injector temperature: 220 °C, TCD detector temperature and FID detector temperature: 220 °C. For determining the gaseous product a separate reaction was carried out and product was analysed after 24 h of vis-irradiation by injecting 20 μL sample in GC-TCD and GC-FID.
Blank reactions were performed for ensuring that originated methanol was photoreduction product of CO2 and not originated due to degradation of organic materials. First blank reaction was carried out in identical conditions except no catalyst was added. In the second blank reaction, catalyst and same solvent system was used except reaction was placed in dark. An additional blank experiment was also carried out in Vis-irradiation containing catalyst but purging N2 rather than CO2. No any product was detected in all the blank reactions. Further, to checking effect of solvent system we have carried out reaction in another aprotic solvent acetonitrile and water system in identical conditions. The yield of methanol in this system was found to be comparable to DMF (Fig. S2†).
Acknowledgements
We would like to thanks Director IIP for granting permission to publish these findings. PK and AK are also thankful to UGC and CSIR, New Delhi for providing fellowships. Analytical department is kindly acknowledged for analysis of samples.
Notes and references
-
(a) G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed;
(b) E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC;
(c) M. M. Halmann and M. Steinberg, Greenhouse Gas Carbon Dioxide Mitigation Science and Technology, Lewis Publishers, 1999 Search PubMed;
(d) M. Aresta, Carbon dioxide reduction and use as a chemical feedstock, ed. W. B. Tolman, Wiley-VCH, Weinheim, Germany, 2006, pp. 1–41 Search PubMed;
(e) M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC;
(f) D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
-
(a) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed;
(b) S. N. Habisreutinger, L. S. Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed;
(c) A. Inagakia and M. Akita, Coord. Chem. Rev., 2010, 254, 1220–1239 CrossRef PubMed;
(d) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed;
(e) W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed;
(f) R. Reithmeier, C. Bruckmeier and B. Rieger, Catalysts, 2012, 2, 544–571 CrossRef CAS PubMed.
- T. Inoue, A. Fujimshima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS PubMed.
-
(a) Q. Zhang, Y. Li, E. A. Ackerman, M. G. Josifovska and H. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS PubMed;
(b) R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS PubMed;
(c) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC;
(d) T.-V. Nguyen, J. C. S. Wua and C.-H. Chiou, Catal. Commun., 2008, 9, 2073–2076 CrossRef CAS PubMed;
(e) C. Wang, X.-X. Ma, J. Li, L. Xu and F.-x. Zhang, J. Mol. Catal. A: Chem., 2012, 363–364, 108–114 CrossRef CAS PubMed;
(f) X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
-
(a) Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS PubMed;
(b) A. Kubacka, M. F. Garcia and G. Colon, Chem. Rev., 2012, 112, 1555 CrossRef CAS PubMed;
(c) M. R. Hoffmann, J. A. Moss and M. M. Baum, Dalton Trans., 2011, 40, 5151–5158 RSC;
(d) A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC.
- S. Naval, A. Dhakshinamoorthy, M. lvaro and H. Garcia, ChemSusChem, 2013, 6, 562–577 CrossRef PubMed.
-
(a) Y. Feng, N. Feng, G. Zhang and G. Du, CrystEngComm, 2014, 16, 214–222 RSC;
(b) Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2012, 6, 9777–9789 CrossRef CAS PubMed.
-
(a) A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC;
(b) T. Yui, Y. Tamaki, K. Sekizawa and O. Ishitani, in Photocatalysis, ed. C. A. Bignozzi, 2011, vol. 303, pp. 151–184 Search PubMed.
-
(a) I.-H. Tseng, J. C. S. Wu and H.-Y. Chou, J. Catal., 2004, 221, 432–440 CrossRef CAS PubMed;
(b) L. Liu, F. Gao, H. Zhao and Y. Li, App. Catal., B, 2013, 134–135, 349–358 CrossRef CAS PubMed.
-
(a) L. Liu, D. T. Pitts, H. Zhao, C. Zhao and Y. Li, App. Catal., A, 2013, 7, 474–482 CrossRef PubMed;
(b) A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
-
(a) N. Sasirekha, S. J. S. Basha and K. Shanthi, App. Catal., B, 2006, 62, 169–180 CrossRef CAS PubMed;
(b) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
-
(a) M. D. Sampson, J. D. Froehlich, J. M. Smieja, E. E. Benson, I. D. Sharpbc and C. P. Kubiak, Energy Environ. Sci., 2013, 6, 3748–3755 RSC;
(b) H. Takeda, H. Koizumi, K. Okamotoa and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC;
(c) S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fre, P. Rubino, C. Chone, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS PubMed;
(d) S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
-
(a) F. Puntoriero, G. L. Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725–4727 RSC;
(b) P. Voyame, K. E. Toghill, M. A. Mendez and H. H. Girault, Inorg. Chem., 2013, 52, 10949–10957 CrossRef CAS PubMed;
(c) H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, J. Photochem. Photobiol., A, 1996, 96, 171–174 CrossRef CAS;
(d) J. Huang, D. Stockwell, Z. Huang, D. L. Mohler and T. Lian, J. Am. Chem. Soc., 2008, 130, 5632–5633 CrossRef CAS PubMed.
-
(a) S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed;
(b) J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed;
(c) S. Metz and S. Bernhard, Chem. Commun., 2010, 46, 7551–7553 RSC;
(d) L. L. Tinker and S. Bernhard, Inorg. Chem., 2009, 48, 10507–10511 CrossRef CAS PubMed.
-
(a) Z.-H. Zhao, J.-M. Fan and Z.-Z. Wang, J. Cleaner Prod., 2007, 15, 1894–1897 CrossRef PubMed;
(b) J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. M. Ferrero, E. Palomares, J. Ko, M. Gratzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358–8362 CrossRef CAS PubMed;
(c) J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
-
(a) Z. Zhao, J. Fan, M. Xie and Z. Wang, J. Cleaner Prod., 2009, 17, 1025–1029 CrossRef CAS PubMed;
(b) V. Iliev, J. Photochem. Photobiol., A, 2002, 151, 195–199 CrossRef CAS;
(c) V. Iliev and A. Mihaylova, J. Photochem. Photobiol., A, 2002, 149, 23–30 CrossRef CAS.
- P. Kumar, A. Kumar, B. Sridhar, B. Sain, S. S. Ray and S. L. Jain, Chem. Eur. J., 2014, 20, 6154–6161 CrossRef CAS PubMed.
-
(a) P. Kumar, S. Kumar, S. Cordier, S. Paofai, R. Boukherroub and S. L. Jain, RSC Adv., 2014, 4, 10420 RSC;
(b) P. Kumar, A. Bansiwal, N. Labhsetwar and S. L. Jain, Green Chem., 2015, 17, 1605–1609 RSC;
(c) P. Kumar, B. Sain and S. L. Jain, J. Mater. Chem. A, 2014, 2, 11246–11253 RSC.
- D. Valecchha, S. Lokhande, M. Klementova, J. Subrt, S. Rayalu and N. Labhsetwar, J. Mater. Chem., 2011, 21, 3718–3725 RSC.
-
(a) J. Mack and M. J. Stillman, Coord. Chem. Rev., 2001, 219, 993–1032 CrossRef;
(b) S. Bon, L. Valentini and J. M. Kenny, Chem. Phys. Lett., 2010, 494, 264–268 CrossRef PubMed;
(c) R. R. Salinas-Guzman, J. L. Guzman-Mar, L. Hinojosa-Reyes, J. M. Peralta-Hernandez and A. Hernandez-Ramirez, J. Sol-Gel Sci. Technol., 2010, 54, 1–7 CrossRef CAS PubMed.
-
(a) P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS PubMed;
(b) L. J. Seok and C. S. Churl, Mater. Lett., 2004, 58, 390–393 CrossRef.
-
(a) W. Liu, L. Feng, C. Zhang, H. Yang, J. Guo, X. Liu, X. Zhang and Y. Yang, J. Mater. Chem. A, 2013, 1, 6942–6948 RSC;
(b) B. Q. Yuan, H. H. Duan, L. L. Li, Z. X. Li, W. T. Duan, L. S. Zhang, W. G. Song and C. H. Yan, Adv. Mater., 2010, 22, 1475–1478 CrossRef PubMed.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CrossRef CAS.
-
(a) O. Ohno, N. Ishikawa, H. Matsuzawa, Y. Kaizu and H. Kobayashi, J. Phys. Chem., 1989, 93, 1713–1718 CrossRef CAS;
(b) C. Ercolani, A. M. Paoletti, G. Pennesi and G. Rossi, J. Chem. Soc., Dalton Trans., 1990, 1971 RSC.
-
(a) A. Ogunsipe and T. Nyokong, Photochem. Photobiol. Sci., 2005, 4, 510–516 RSC;
(b) A. Ogunsipe and T. Nyokong, J. Photochem. Photobiol., A, 2005, 173, 211–220 CrossRef CAS PubMed.
-
(a) D. Zhang, H. Fu, L. Shi, C. Pan, Q. Li, Y. Chu and W. Yu, Inorg. Chem., 2007, 46, 2446–2451 CrossRef CAS PubMed;
(b) C. Ho, C. Y. Jimmy, K. Tszyan, C. M. Angelo and L. Sukyin, Chem. Mater., 2005, 17, 4514–4522 CrossRef CAS.
-
(a) A. B. Sorokin and A. Tuel, New J. Chem., 1999, 23, 473 RSC;
(b) A. B. Sorokin, S. Mangematin and C. Pergrale, J. Mol. Catal. A: Chem., 2002, 182–183, 267 CrossRef CAS.
- P. Pavaskar, S. Chodankar and A. Salker, Eur. J. Chem., 2011, 2, 416–419 CrossRef CAS PubMed.
-
(a) J. Yang, L. Lukashuk, H. Li, K. Fottinger and G. Rupprechter, Catal. Lett., 2014, 144, 403–412 CrossRef CAS PubMed;
(b) K. M. S. Khalil, J. Colloid Interface Sci., 2007, 307, 172–180 CrossRef CAS PubMed.
-
(a) D. R. A. El-Hafiz, M. A. Ebiad, R. A. Elsalamony and L. S. Mohamed, RSC Adv., 2015, 5, 4292–4303 RSC;
(b) I. M. Hunga, D. T. Hunga, K. Z. Funga and M. H. Hon, J. Eur. Ceram. Soc., 2006, 26, 2627–2632 CrossRef PubMed.
-
(a) N. Masilela and T. Nyokong, Dyes Pigm., 2011, 91, 164–169 CrossRef CAS PubMed;
(b) T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS PubMed;
(c) The Handbook of Porphyrin Science, Academic Press, World Scientific, New York, Singapore, 2010, vol. 7, pp. 247–349 Search PubMed.
-
(a) I. S. Mokhoshi, N. Kuznetsova and T. Nyokong, J. Potochem. Photobiol., A, 2001, 140, 215–222 CrossRef;
(b) K. Ozoemen, N. Kuznetsova and T. Nyokong, J. Photochem. Photobiol., A, 2001, 139, 217–222 CrossRef.
-
(a) J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 3200–3207 CrossRef CAS PubMed;
(b) B. Probst, A. Rodenberg, M. Guttentag, P. Hamm and R. Alberto, Inorg. Chem., 2010, 49, 6453–6460 CrossRef CAS PubMed;
(c) C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161–2166 CrossRef CAS.
-
(a) W. J. Youngblood, S. H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed;
(b) G. Centi and S. Perathoner, ChemSusChem, 2010, 3, 195–208 CrossRef CAS PubMed;
(c) K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., 2012, 124, 8132–8135 CrossRef PubMed.
-
(a) P. Barrett, C. Dent and P. Linstead, J. Chem. Soc., 1936, 1719 RSC;
(b) W. J. Kroenke and M. E. Kenney, Inorg. Chem., 1964, 3, 251–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and surface characterization data of rGO-immobilized catalyst. See DOI: 10.1039/c5ra06449j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.