
Preparation and characterization of new poly(dimethylsiloxane) membrane series via a ‘cross-linking’ reaction using monomolecular trichloro(alkyl)silane of different alkyl chain and type
Abstract
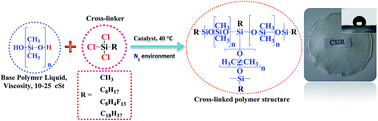
Poly(dimethylsiloxane) (PDMS) membrane, which is generally prepared by cross-linking a hydroxyl-terminated-poly(dimethylsiloxane) liquid with a polymethylhydrosiloxane cross-linker through a condensation reaction between the hydroxyl end groups and hydride groups liberating hydrogen, is one of the most studied polymeric membranes for the separation of gases and liquids. Herein, a new PDMS membrane series prepared by direct cross-linking hydroxyl terminated polydimethylsiloxane pre-polymer liquid with RSiCl3 molecules of different types in n-heptane solvent under a nitrogen environment were studied, wherein the alkyl chain, R, was varied as methyl CH3, octyl C8H17, perfluorooctyl C8H4F13, and octadecyl C18H37. For each membrane series, the amount of cross-linker to pre-polymer was varied as 13 : 87, 33 : 67 and 50 : 50 (w/w) to compare the membranes at different cross-linking densities. The cross-linked network structure of the membrane comprised dimethylsiloxane network structures of two cross-links and alkylsiloxane network structure of two or three cross-links. The changes in the structure and properties of the membranes depending on the concentration and type of trichloro(alkyl)silane used were observed by XRD, SEM, TEM, SANS, TGA, DSC, 29Si NMR, IR, cross-linking density, contact angle (water) and ethanol–water separation performance measurements. The membranes with an optimal amount of trichloro(alkyl)silane cross-linker of approximately 10–30% (w/w) showed better separation performance than the other reported conventional PDMS membranes in terms of the ethanol selectivity and flux from an aqueous feed containing 5% (w/w) ethanol. Among the membranes prepared, the membranes cross-linked with perfluorooctylsiloxane/octadecylsiloxane showed better separation performance than those membranes cross-linked with methylsiloxane/octylsiloxane.
 Please wait while we load your content...
Please wait while we load your content...

